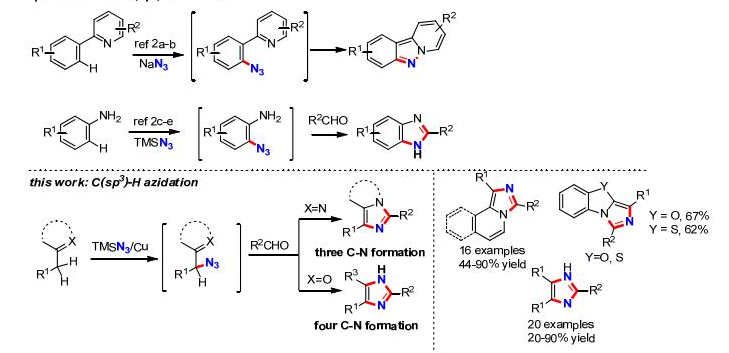
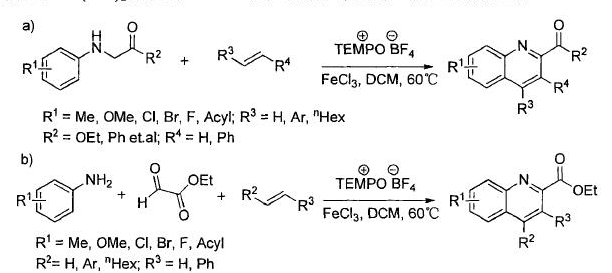
非活化苄基C-H键的叠氮化反应毕业论文
2020-07-01 20:49:07
摘 要
在有机合成、化学生物学和药物发现中,C-H键分子发挥着重要作用,然而,在合成反应库中,却明显缺乏将脂肪族C-H键直接转变为烷基叠氮化物的反应。利用有机分子中C-H键的普遍性和叠氮官能团的多功能性,这种转化可以在各种学科中广泛应用。活化的C-H键代表了脂肪族C-H叠氮化的吸引力的策略,如在活化惰性脂肪族C-H键方面它克服了采用常规有机金属带来的许多缺点。新的C-H叠氮化方法可以通过将氢原子取代的自由基C-H活化与合适的叠氮化物转移试剂组合来实现。从这个角度来看,我们考察了活化的C-H叠氮化的历史,并总结了该领域近期的一些显着进展。
迄今为止,所有的自由基C-H叠氮化物都遵循常规的方法,包括初始的自由基C-H提取步骤和随后的叠氮化物转移到初始以碳为中心的自由基。与先前的方法相比,这一观点的独特之处是在C-H叠氮化反应中充分利用了过渡金属催化剂的优点,它们“驯服”了叠氮化物自由基,并在更为温和的情况下高效进行的反应,并提供更宽的底物范围和更高的区域选择性和立体选择性。
关键词:C-H活化 自由基 叠氮化 催化 有机叠氮化物
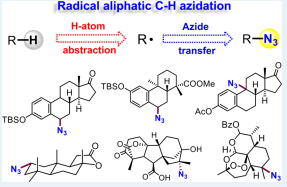
Taming Azide Radicals for Catalytic C−H Azidation
Abstract
Reactions that directly transform aliphatic C−H bonds into alkyl azides are noticeably lacking in the repertoire of synthetic reactions, despite the importance of molecules containing C−N3 bonds in organic synthesis, chemical biology, and drug discovery. Harnessing the ubiquity of C−H bonds in organic molecules and the versatility of the azide functional group, such transformations could have broad applications in various disciplines. Radical C−H activation represents an appealing strategy to achieve aliphatic C−H azidation, as it overcomes many drawbacks of conventional organometallic approaches in activating inert aliphatic C−H bonds. Novel C−H azidation methodologies could be realized by combining radical C−H activation via hydrogen atom abstraction with suitable azide-transfer reagents. In this perspective, we survey the history of radical C−H azidation and summarize several significant recent advances in the field.
All radical C−H azidations to date follow a general approach comprising an initial radical C−H abstraction step and a subsequent azide transfer to the incipient carbon-centered radicals. A particular focus of this perspective is on the beneficial effects of using transition-metal catalysts in C−H azidation reactions, which have “tamed” azide radicals and led to reactions that proceed efficiently under much milder conditions and provide broader substrate scope and higher regioselectivities and stereoselectivities, compared to previous approaches.
Key Words: C−H activation, radical, azidation, catalysis, organic azides
目 录
摘 要 I
Abstract II
第一章 文 献 综 述 1
1.1引言 1
1.2叠氮化合物 1
1.3脂肪族C-H键自由基的连接 2
1.3.1叠氮基团的热力学性质 2
1.3.2有机氢原子取代基介导的脂肪族C-H叠氮化 3
1.4碳氢活化叠氮化 8
1.4.1 通过导向基构建叠氮化方法 8
1. 4. 2 铜催化的叠氮化方法 8
1.4.3 银催化叠氮化反应 9
1.5 N-酰基四氢异喹啉的碳氢修饰 9
1.5.1氧合铵盐(TEMPO Oxoammonium Salt)氧化体系 10
1.5.2喹啉的碳氢活化合成 11
1.6.结语 11
第二章 实验内容 12
2.1 仪器与试剂 12
2.1.1实验试剂 12
2.1.2实验仪器 13
2.2 实验部分 14
2.2.1氧化剂的制备 14
2.2.2反应条件筛选及优化 15
2.3结果及讨论 21
2.4化合物结构表征 22
2.5化合物代表性谱图 22
第三章 实验总结和展望 23
3.1实验总结 23
3.2实验展望 23
参考文献 24
致谢 25
第一章 文 献 综 述
1.1引言
在有机合成、化学生物学和药物发现中,C-H键分子发挥着重要作用,然而,在合成反应库中,却明显缺乏将脂肪族C-H键直接转变为烷基叠氮化物的反应。利用有机分子中C-H键的普遍性和叠氮官能团的多功能性,这种转化可以在各种学科中广泛应用。活化的C-H键代表了脂肪族C-H叠氮化的吸引力的策略,如在活化惰性脂肪族C-H键方面它克服了采用常规有机金属带来的许多缺点。新的C-H叠氮化方法可以通过将氢原子取代的自由基C-H活化与合适的叠氮化物转移试剂组合来实现。从这个角度来看,我们考察了活化的C-H叠氮化的历史,并总结了该领域近期的一些显着进展。
迄今为止,所有的自由基C-H叠氮化物都遵循常规的方法,包括初始的自由基C-H提取步骤和随后的叠氮化物转移到初始以碳为中心的自由基。与先前的方法相比,这一观点的独特之处是在C-H叠氮化反应中充分利用了过渡金属催化剂的优点,它们“驯服”了叠氮化物自由基,并在更为温和的情况下高效进行的反应,并提供更宽的底物范围和更高的区域选择性和立体选择性。
1.2叠氮化合物
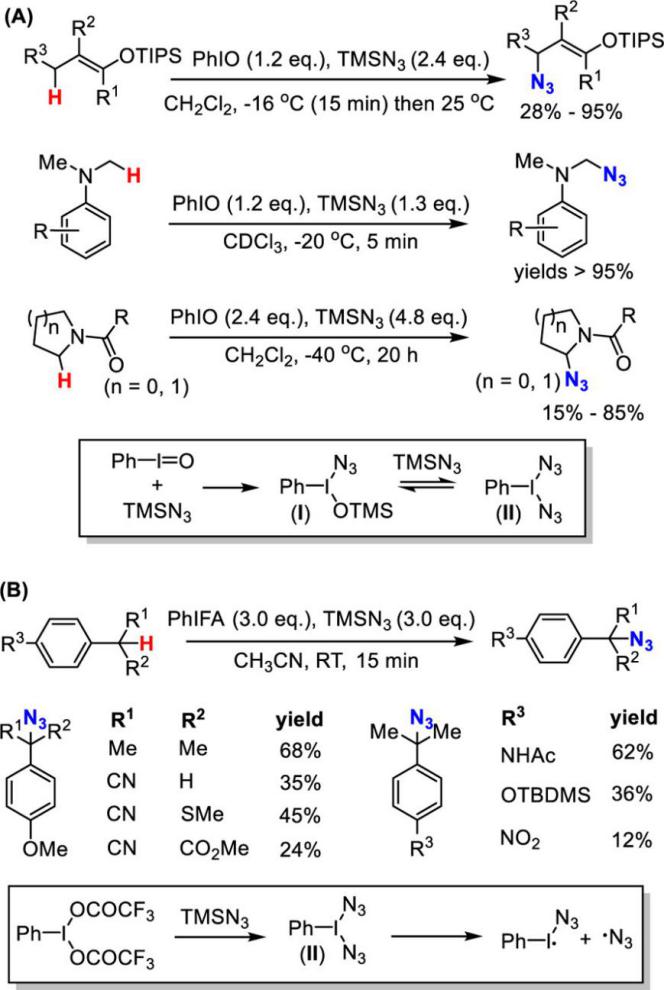
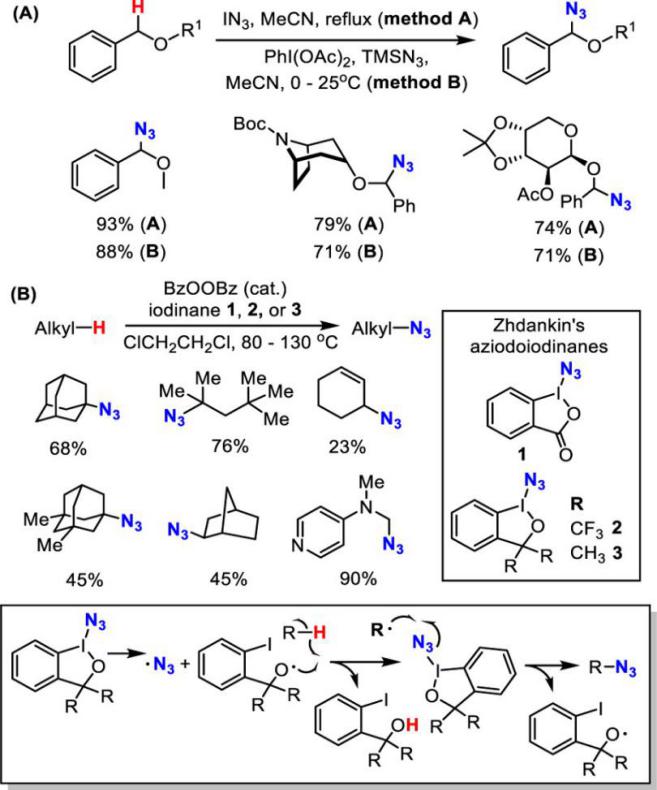
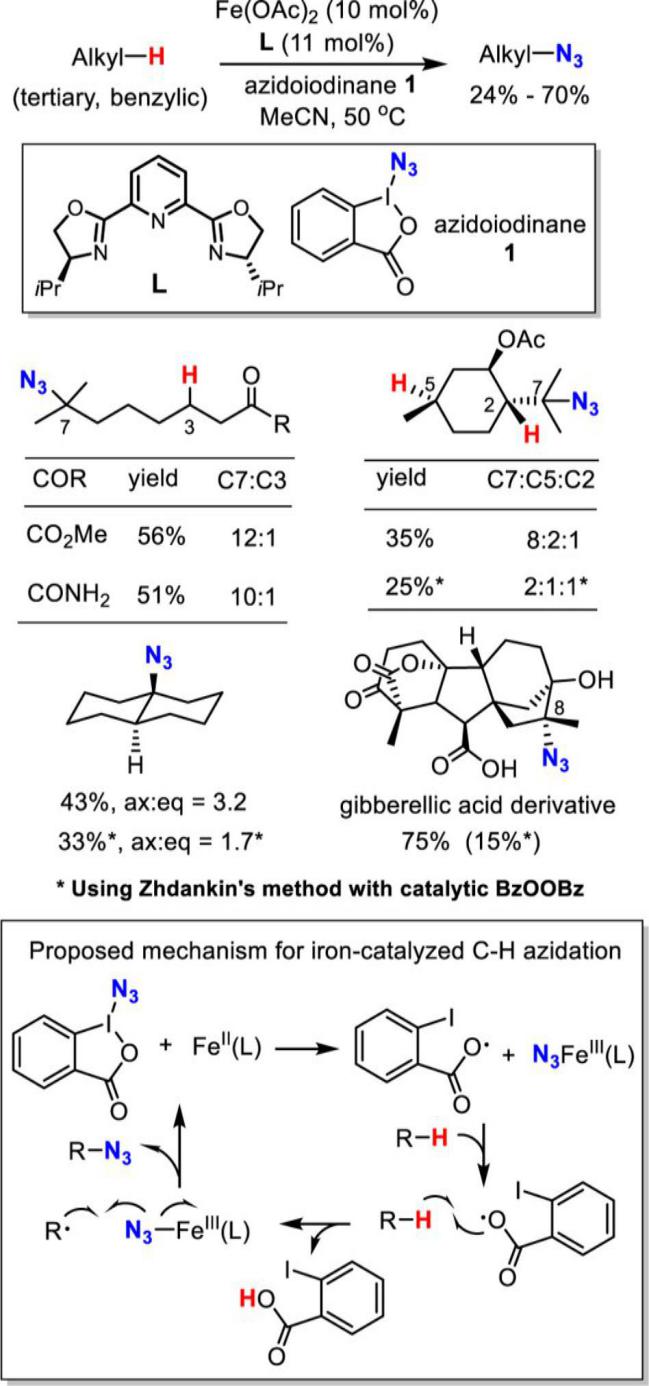
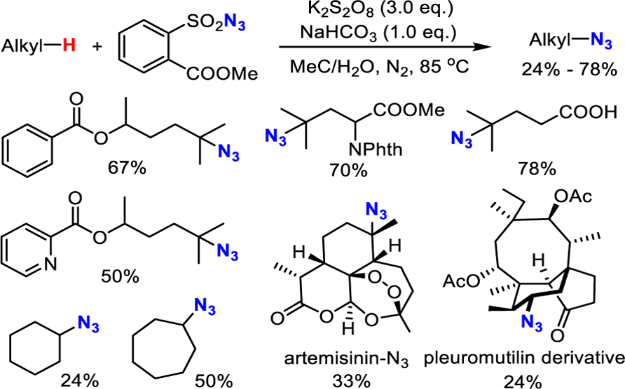
已经过去的十年来,通过C-H活化构建C-N键已经取得了巨大的成就。 最近,通过使用过渡金属催化(Cu,Pd或Rh)或高价碘试剂的C-H活化获得芳基叠氮化物已经取得显著进展[4]。相比之下,明显缺乏通过C-H直接活化合成脂肪族叠氮化合物的方法。脂肪族C-H叠氮化反应的一个主要挑战在于这种强非极性键的动力学顽抗。二甲基亚砜(DMSO)中特别低的酸度(pKa= 43-59))[5]和脂肪族C-H键的弱配位能力使它们很难用有机金属方法活化,如氧化加成和亲电活化。虽然已知几种有机金属体系如Shilov化学可以使未活化的脂肪族C-H键功能化[6],但他们通常需要强制条件并且主要适用于简单的碳氢化合物。通常使用导向基团策略,扩大底物范围并实现高选择性和温和条件,其中预先安装具有孤对电子和/或π-系统的官能团以帮助分子与金属中心的配位。引入基团的结合限制了基底的结构变化,并且需要额外的步骤来安装和去除引导基团降低整体合成效率。然而,这些限制不存在于自由基的C-H官能化中,其通过氢原子取代活化脂族C-H键,并且因此C-H反应性主要取决于均聚键解离能(二苯醚)[8]。已知许多有机和过渡金属体系通过在温和条件下具有良好选择性和官能团耐受性的氢原子转移而高效活化脂肪族C-H键[8,9]。可以想象的是,将这些H原子取代基与合适的叠氮化物转移剂结合,借此提供实现脂肪族C-H叠氮化的常见策略(参见方案1)。从这个角度来看,我们从历史角度观察脂肪族C-H叠氮化,并总结最近的进展。我们将特别关注如何部署金属催化剂以提高反应效率并提升C-H叠氮化反应中所需的区域选择性和立体选择性。
相关图片展示: