动力学拆分对α-羟基和α-氨基苯乙酸的对映选择性C-H键烯烃化反应外文翻译资料
2023-01-04 11:08:34
杭 州 师 范 大 学
本科生毕业设计(论文)文献翻译
|
题 目 |
动力学拆分对alpha;-羟基和alpha;-氨基苯乙酸的对映选择性C-H键烯烃化反应 |
||
|
|
|||
摘要:近十年来,对映选择性的C-H活化反应的研究取得了重大进展。但是,要求存在两种化学上相同的前手性C-H键使得其在范围上具有一定的局限性。本次报道的是动力学拆分的第一个例子通过钯(II)催化对映选择性C-H活化和C-C键形成,从而显著扩大了对映选择性C-H活化反应的范围。
对映选择性C-H活化反应的发展在催化和有机合成中是一个重要且具有挑战性的任务,因为它们为不对称合成提供了新的断开。在各种方法中,前手性C-H键的钯催化的去对称化作为一种有有效的途径,它可以产生大量的对映选择性碳-碳和碳-杂原子键的形成反应。然而,去对称化反应仅适用于含有两个前手性C-H键的底物,因此限制了该方法可以获得的结构多样性。 例如,先前报道的(图1a)对映选择性钯(II)-催化的二苯基乙酸的C-H烯烃化反应通过使用单-N-保护氨基酸(MPAA)配体不能用于制备重要的扁桃酸和苯基甘氨酸的手性衍生物。
图1a
对映体纯的alpha;-羟基和alpha;-氨基苯乙酸(也称为扁桃酸和苯基甘氨酸)分别是许多药物和生物活性化合物如抗生素头孢噻吩,头孢氨苄和万古霉素中发现的重要结构基序。对映体富含的扁桃酸和苯基甘氨酸衍生物既可用作催化剂,也可用作有机合成中的手性结构单元。因此,这两个族的化合物的不对称合成受到了很多关注。手性环氧化物的苄基C-H键不对称羟基化的早期实例和最近使用PdII/MPAA催化剂的对映选择性C-H碘化的反应证明了实现通过C-H羟基化和碘化实现动力学拆分的可行性。在此,我们报道了通过动力学拆分的钯(II)催化的alpha;-羟基和alpha;-氨基苯乙酸的对映选择性C-H烯化反应,得到对映体富集的烯烃化扁桃酸和苯基甘氨酸(图1b)。使用手性MPAA配体进一步对剩余原料进行对映选择性烯烃化反应,以相反的构型,使对映体具有高的对映体纯度。值得注意的是,这些手性分子不能通过前手性碳氢键的去甲基化或其他不对称方法得到。据我们所知,这个反应是第一个通过碳氢键激活和形成碳氢键来进行动力学拆分的例子。
图1b
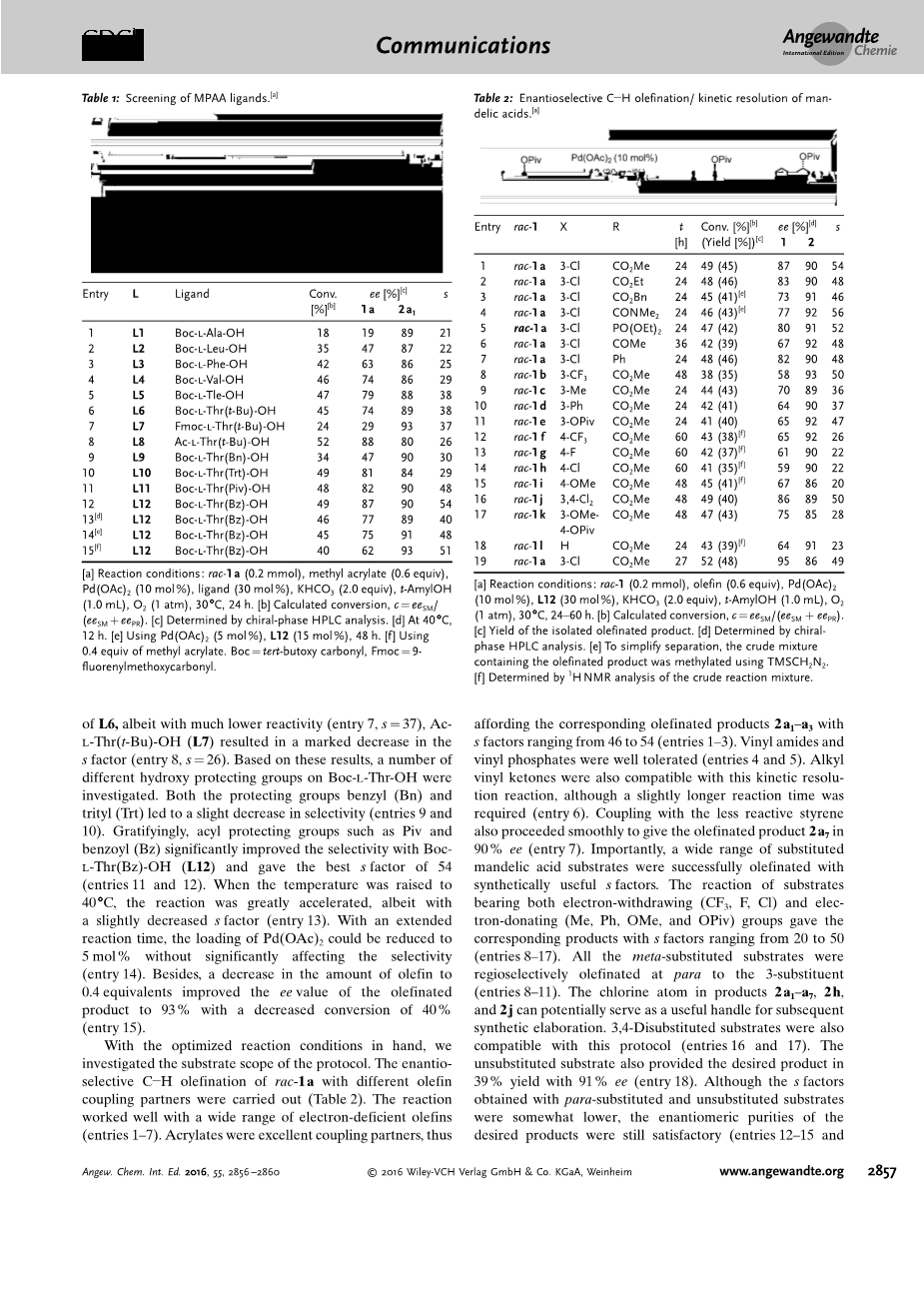
本研究以丙烯酸甲酯为偶联剂,探讨外消旋异戊酰(Piv)保护的3-氯扁桃酸racl-1a的对映选择性C-H烯烃化/动力学拆分(表1-保护组筛选的支持信息)。研究发现,在Boc-l-Ala-OH(L1)作为配体存在的情况下,ac-1a在有氧条件下对映选择性C-H烯化反应得到的产物2a1在18%的转化率下具有89%的ee,因此对应的选择性因子(s)为21 (条目1)。在这一令人鼓舞的结果的鼓舞下,我们筛选了多种具有不同侧链的Boc保护氨基酸配体(L1-L6)。随着侧链上空间体积的增加,这些因素逐渐得到改善(条目1-6)。s因子随着侧链空间体积的增大而逐渐改善(条目1-6)。具体来说,Boc-l-Tle -OH (L5)和Boc-l-Thr(t-Bu)-OH (L6)给出了38个优越的s因子(条目5和6)。由于苏氨酸上的羟基(H-1-Thr-OH)可以为结构修饰提供有价值的手段,因此选择该氨基酸作为配体骨架用于进一步调整。首先,我们研究了不同N-保护基团的作用。虽然Fmoc-1-Thr(t-Bu)-OH(L8)提供与L6相似的因子,尽管具有低得多的反应性(条目7,s = 37),Ac-1-Thr(t-Bu) - OH(L7)导致因子显著减少(条目8,s=26)。基于这些结果,我们研究了Boc-1-Thr-OH上的许多不同的羟基保护基团。保护基团苄基(Bn)和三苯甲基(Trt)均导致选择性略微降低(条目9和10)。令人高兴的是,酰基保护基团如Piv和苯甲酰(Bz)显着提高了Boc-l-Thr(Bz)-OH(L12)的选择性,并得到了最佳的因子54(条目11和12)。当温度升至40℃时,反应大大加速,尽管s因子略微降低(条目13)。随着反应时间的延长,Pd(OAc)2的负载量可降低至5mol%而不会显着影响选择性(条目14)。此外,将烯烃量减少至0.4当量使烯烃化产物的ee值提高至93%,转化率降低40%(条目15)。
|
Entry |
L |
Ligand |
Conv.[%] |
ee[%] 1a 2a |
s |
|
|
1 |
L1 |
Boc-L-Ala-OH |
18 |
19 |
89 |
21 |
|
2 |
L2 |
Boc-L-Leu-OH |
35 |
47 |
87 |
22 |
|
3 |
L3 |
Boc-L-Phe-OH |
42 |
63 |
86 |
25 |
|
4 |
L4 |
Boc-L-Val-OH |
46 |
74 |
86 |
29 |
|
5 |
L5 |
Boc-L-Tle-OH |
47 |
79 |
88 |
38 |
|
6 |
L6 |
Boc-L-Thr(t-Bu)-OH |
45 |
74 |
89 |
38 |
|
7 |
L7 |
Fmoc-L-Thr(t-Bu)-OH |
24 |
29 |
93 |
37 |
|
8 |
L8 |
Ac-L-Thr(t-Bu)-OH |
52 |
88 |
80 |
26 |
|
9 |
L9 |
Boc-L-Thr(Bn)-OH |
34 |
47 |
90 |
30 |
|
10 |
L10 |
Boc-L-Thr(Trt)-OH |
49 |
81 |
84 |
29 |
|
11 |
L11 |
Boc-L-Thr(Piv)-OH |
48 |
82 |
90 |
48 |
|
12 |
L12 |
Boc-L-Thr(Bz)-OH |
49 |
87 |
90 |
54 |
|
13 |
L12 |
Boc-L-Thr(Bz)-OH |
46 |
77 |
89 |
40 |
|
14 |
L12 |
Boc-L-Thr(Bz)-OH |
45 |
75 |
91 |
48 |
|
15 |
L12 |
Boc-L-Thr(Bz)-OH |
40 |
62 |
93 |
51 |
表 1
在优化的反应条件下,我们研究了协议的底物范围。采用不同烯烃偶联剂对映选择性C-H烯烃化反应(表2)。该反应在大量缺电子烯烃上效果良好(条目1-7)。丙烯酸酯是极好的偶联剂,因此提供了相应的s因子在46-54之间的烯化产物2a1-a3(条目1 - 3)。乙烯基酰胺和乙烯基磷酸盐耐受性良好(条目4和条目5)。烷基乙烯基酮也与这种动力学拆分反应相容,尽管需要稍长的反应时间(条目6)。与活性较低的苯乙烯进行偶联,也可以在90%的ee下顺利地得到烯化产物2a7(条目7)。重要的是,广泛的取代扁桃酸底物成功地与综合有用的s因子烯烃化。同时具有吸电子(CF3,F,Cl)和供电子(Me, Ph, OMe, OPiv)基团的底物的反应得到了s因子为20-50的相应产物(条目8-17)。所有的间取代基在3-取代基的对位上选择性地烯烃化(条目8-11)。产物2a1-a7、2h、2j中的氯原子可以作为后续合成精化的有用手段。3,4-二取代的底物也与该方案相容(条目16和17)。未取代的底物也以39%的收率提供所需产物,具有91%ee(条目18)。尽管用对位取代和未取代的底物获得的s因子稍低,但所需产物的对映体纯度仍然令人满意(条目12-15和18)。值得注意的是,当转化率略高于50%时,可以获得原料的高对映体纯度。 例如,在52%转化率下,在95%ee中回收(R)-3-氯扁桃酸(1a)(条目19)。
剩余内容已隐藏,支付完成后下载完整资料

英语原文共 5 页,剩余内容已隐藏,支付完成后下载完整资料
资料编号:[278798],资料为PDF文档或Word文档,PDF文档可免费转换为Word



