二维2H-MoTe2材料的第一性原理研究毕业论文
2021-12-09 17:20:32
论文总字数:21513字
摘 要
二十一世纪是一个可持续发展的世纪,在各个领域对于先进材料的研究都进入了一个快速发展的阶段。考虑到经济可持续发展以及社会可持续发展,为了若干年后我们的子孙后代不会因为环境恶化而失去家园,对材料的研究就要首先考虑节能型以及环境友好型。
二维单层的(Two dimensional)过渡金属硫系化合物(TMDs),由于其具有直接带隙大,体形超薄,具有良好的电子迁移率和通用的电子结构而备受关注。他们在未来的电子、光电、光伏、相变和光催化等领域中都显示出了巨大的潜力。要明确它们在这些领域上的适用性,了解它们的基本电子结构和结构稳定性是先决条件。与石墨烯不同,TMDs存在多个多晶型物(例如2H、1T和3R等)。每种晶型都有其独特的性质,这给了我们很大的选择空间,可以通过选择不同的材料相互配合组成异质结或是通过调制能带结构来达到我们的要求。因此,为了能够实现这个目标,首先就需要对材料的电子结构以及晶体结构有一个全面的了解。
密度泛函理论(DFT)的研究以及计算机技术的发展催生了计算材料学,通过在计算机上进行模拟既可以大大减少实验室实验的资源消耗,又可以很大地缩短材料的研究周期,是一种符合二十一世纪发展规律的,方便高效快捷的研究手段。
本文主要运用Materials studio 软件以及CASTEP程序包,对二维2H-MoTe2的晶体结构、能带结构进行研究
主要研究内容包括:
1. 通过建立体相2H-MoTe2、单层2H-MoTe2以及多层的2H-MoTe2的晶体模型,运用CASTEP程序包对所建立的结构进行优化,分析研究随着层数的变化材料的晶体结构、键长、键角、层间距的变化规律。
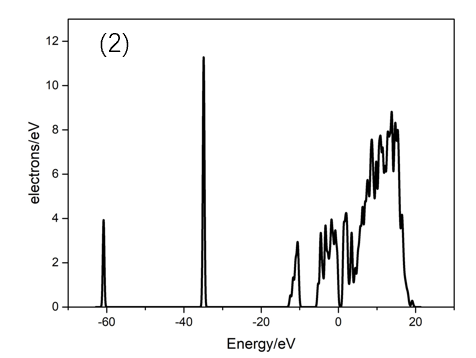
2. 对结构优化后的单层2H-MoTe2、多层2H-MoTe2的电子结构进行分析计算,分析研究随着层数的变化能带结构、带隙大小的变化规律。同时,对不同层数2H-MoTe2的态密度进行了计算。
关键词:2H-MoTe2,二维材料,密度泛函理论,第一性原理研究
Abstract
The 21st century is a century of sustainable development, and the research on advanced materials in various fields has entered a stage of rapid development. In order to achieve sustainable economic development and let future generations enjoy green water and green mountains, the research of materials should first consider energy-saving and environment-friendly.
Single layer Two-dimensional (2D) Transition Metal Dichalcogenides (TMDs) have attracted much attention due to their large direct band gap, ultra-thin shape, good electron mobility and general electronic structure. They have shown great potential in the fields of electron, photoelectricity, photovoltaic, phase transition and photocatalysis in the future. To understand their applicability in these fields, understanding their basic electronic structure and structural stability is a prerequisite. Unlike graphene, there are many polymorphic forms of the same TMDs (such as 2H and 1T or 3R). Each crystal has its unique properties, which gives us a lot of choice space. We can meet our requirements by choosing different materials to form heterojunction or by modulating energy band structure. Therefore, in order to achieve this goal, we need to have a comprehensive understanding of the electronic structure and crystal structure of materials.
The research of Density functional theory (DFT) and the development of computer technology give birth to computational materials. Simulation experiments on computers can greatly reduce the resource consumption of laboratory experiments, and also greatly shorten the research cycle of a material. It is a convenient, efficient and fast research method that conforms to the development law of the 21st century.
In this paper, the crystal structure and band structure of low dimensional 2H-MoTe2 are studied by using materials Studio software and CASTEP package
The main research contents include:
1. By building the crystal models of solid phase 2H-MoTe2, single layer 2H-MoTe2 and multi-layer 2H-MoTe2, using the CASTEP package to optimize the structure, analyze and study the change rules of crystal structure, bond length, bond angle and layer spacing with the change of layers.
2. The energy band structure and density of state of single-layer 2H-MoTe2 and multi-layer 2H-MoTe2 after structure optimization are analyzed and calculated, and the change rule of energy band structure and band gap size with the change of the number of layers is analyzed and studied.
Key Words:2H-MoTe2;two-dimensional materials;Density functional theory;first principles research
目录
摘 要 I
Abstract II
第1章 绪论 1
1.1研究背景与研究现状 1
1.2 TMDs材料的性质介绍 1
1.3 本文的研究内容与行文结构 3
第2章 基础理论知识 4
2.1第一性原理计算方法介绍 4
2.2 量子力学基础 4
2.3 密度泛函理论 5
2.3.1 Born-Oppenheimer(绝热近似) 5
2.3.2 Hartree-Fock近似 5
2.3.3 密度泛函理论介绍 6
2.3.4 霍恩伯格—科恩定理(Hohenberg-Kohn定理) 6
2.3.5 科恩—沈吕九方程(Kohn-Sham方程): 7
2.4 交换关联泛函 8
第3章 二维2H-MoTe2的晶体结构及电子结构研究 10
3.1 MoTe2材料介绍 10
3.2 计算软件及参数 10
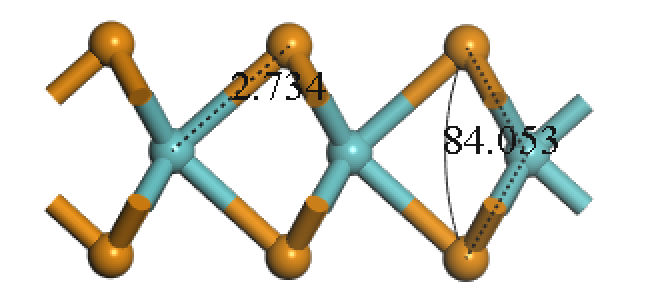
3.3 MoTe2晶体结构 11
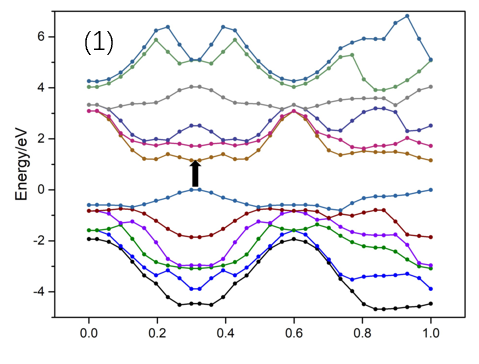
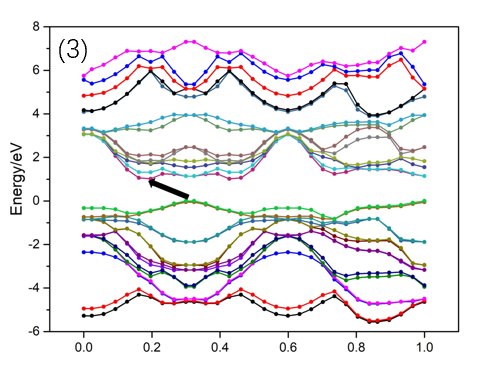
3.4 MoTe2电子结构 13
3.5 本章总结 15
第4章 结论与展望 16
参考文献 17
致 谢 19
第1章 绪论
1.1研究背景与研究现状
2004年英国The University of Manchester的Novoselov 和 Geim 通过利用胶带把石墨剥离成具有单层原子厚度的石墨片---石墨烯。这打破了在物理学研究的很长一段时间内科学家们的概念,证实了有单层的二维材料能在正常条件下稳定存在,他们的这项研究也获得了2010年的诺贝尔奖[1]。它的发现也引领了一个对新物理和广泛应用二维材料的极其活跃的新纪元。石墨烯的结构与单层石墨的结构相同,碳原子有四个价电子,其中每个碳原子与其附近的3个碳原子组成共价键。而四个价电子中有三个以sp2的形式成三个键,而另一个价电子可以分布在整个原子层内,形成一个类似于苯环的离域大π键,所以石墨烯是一种良好的电子和热导体。室温下的电导率可达15000 cm 2V –1s –1,远远超过了已知电导率最高的物质InSb[2]。此外,石墨烯中K点的线性电子色散惹起了许多独特的物理现象,石墨烯的载流子可以被描述为无质量的狄拉克费米子。石墨烯基材料已被广泛用于高速导电器件、光学、润滑剂、混合材料、化学传感器、基因测序和储能器件比如新能源电池等。石墨烯的出现激发了世界范围内对其他二维材料的研究,包括磷烯、六方氮化硼、硅烯、锡烯和过渡金属硫系化合物(TMDs)等。在这些新的二维候选材料中,过渡金属硫系化合物(TMDs)有它独有的特殊的优异性能,比如在场效应晶体管(FET)中具有保证高开关比的大禁带等,近年来受到了学者们广泛的关注[3]。
1.2 TMDs材料的性质介绍
TMDs是一类具有层状MX2结构的材料,其中M代表过渡金属元素,主要包含第四副族到第六副族的元素包括Ti、V、Zr、Hf、Mo、W等,X代表硫系元素包括S、Se或Te。MX2每个原子薄层之间被范德瓦尔斯相互作用这种弱相互作用结合在一起,但在每个层内,一个过渡金属元素(M)的原子和六个硫系元素(X)原子之间形成了强共价键,组成MX6配位八面体。每个MX2层之间形成X-M-X的夹层结构,类似于在两个硫族原子层(X)中间插入一个过渡金属原子层(M)[4]。目前,二硫化钼(MoS2),是一种被研究的最为全面也最好的材料,其结构早在1923年就已被确定。MoS2的结构也为层状结构,层间作用力为范德华力这种弱相互作用,所以最初它主要用作于润滑剂。
根据MX2层的不同的堆积方式及其金属配位形成的不同晶胞结构,TMDs中存在多种多晶型,大致包括1T型、2H型和3R型等,它们分别属于三方晶系、六方晶系以及单斜晶系。其中1T型是以1T’型存在,1T相一般表现为金属相或半金属相如MoTe2和WTe2[5]。而2H相,一般表现为半导体性,如MoS2和WS2。对于同一种材料,不同的晶相稳定性不同,已经进行过的研究表明Mo(W)S2,Mo(W)Se2,MoTe2倾向于形成2H相,而WTe2则倾向于形成1T’相[6]。通过前人科学家的研究表明,每种材料各个相之间在一定的条件下可以互相转变,由于不同的晶相具有不同的电性能,我们可以利用相位工程诱导金属-半导体转变或金属-半金属转变[7]。比如MoTe2材料可以通过高温回火,使其完成从半导体的状态转变为金属态,从而可以改变其电子结构以及电学性质,降低接触电阻,在信息存储设备和光电设备有广泛的潜在应用空间[8]。
请支付后下载全文,论文总字数:21513字
相关图片展示:
您可能感兴趣的文章
- 激光作用下ZrNiSn合金热电材料组成、结构和性能的演化规律开题报告
- 原位生长于碳纤维表面的钒氧化物柔性电极制备开题报告
- 锂硫电池用TixOy-S/HGs复合材料的制备与性能开题报告
- MnO2纳米片修饰ZnO纳米棒阵列的气敏性能研究开题报告
- 基于三维碳基孔结构和电解质协同优化的微型超级电容器文献综述
- 基于C-MEMS工艺的微型混合锂离子电容器构筑及性能开题报告
- 多孔碳负载钼基纳米材料作为高性能析氢电催化剂文献综述
- Cu掺杂ZnxCd1-xS纳米晶的制备与性能研究开题报告
- 用于光伏的III-V族半导体低成本生长外文翻译资料
- 太阳能电池中的GaSb / InGaAs 量子点阱混合结构有源区外文翻译资料



