基于四唑酯构建的金属有机骨架材料及其甲烷存贮性能研究毕业论文
2020-02-19 11:52:02
摘 要
金属有机骨架材料(Metal Organic Frameworks,MOFs)是一种被广泛使用的储气材料和催化剂。MOF材料被如此广泛应用主要是因为其本身具有优异的性能,例如比表面积高和物理化学性质稳定等。不过随着新型材料结构日益多样性导致它们的功能变得多样化,这使得筛选多孔材料变得越来越困难。迄今为止,计算机的发展依旧保持高速,使用计算机模拟计算不仅可以大量筛选不同结构,而且能够省去合成材料所花费的各项成本,奠定了合成理想材料的基础。本篇文章主要是通过计算机分子模拟技术设计新颖的多孔骨架材料并进行优化,然后探究这些材料对甲烷的吸附量,探讨金属骨架材料的甲烷吸附机理。
本文首先通过商业软件Materials Studio进行金属有机骨架材料有机连接体(linkers)的初步设计,然后使用ToBaCCo3.0软件得到1138种具有不同金属中心和拓扑结构的MOF材料,之后采用Materials Studio软件的Forcite模块对已设计的MOF材料进行优化。对得到的MOF材料使用Zeo 软件计算其结构性质例如孔径、比表面积和孔隙率等,从1138种材料中随机抽取50种采用巨正则蒙特卡洛模拟(GCMC)方法模拟他们在温度298K、压力5.8MPa和65MPa下甲烷的吸附量,然后构建出吸附量与三种性质的多元线性回归模型(MLR),用此模型预测出1138种材料的甲烷吸附量,再从其中按最大工作容量降序筛选出吸附性能最优的350种材料,再次用GCMC方法模拟得到这350种材料的甲烷吸附量,并从中筛选出5种性能较好的材料。在常温298K温度条件下和压力在1-80MPa间对这五种材料模拟计算其吸附量,得到五种多孔材料对甲烷的吸附等温线。最后通过这些材料的径向分布函数曲线图和质心密度图可得到材料结构对甲烷吸附机理。
本文通过模拟计算得到,在298K、5.8MPa及65MPa下,fcu拓扑结构以锆金属为中心的MOF材料具有最优的甲烷存贮性能,最优性能材料命名为fcu_(Zr6O8)(TRZ)12_link23 ,通过该材料的径向分布曲线和质心密度图可以发现,甲烷分子在5.8MPa下优先选择吸附在金属中心上,在65MPa下优先吸附在有机连接体上。
关键词:甲烷存贮;金属多孔骨架材料;分子模拟方法;量子力学计算
Abstract
Metal Organic Frameworks (MOFs) are widely used as Materials of gas storage and catalysts. MOF Materials are so used widely mainly because of their excellent properties such as high specific surface area and stable physicochemical properties. However, as the structure of new Materials becomes more diverse, their functions become diversified, which makes it increasingly difficult to screen porous Materials. Nowadays, with the rapid development of computer technology, the use of computer simulation calculation can not only screen a large number of different structures, but also save the cost of synthetic Materials, and lay the foundation for the synthesis of ideal Materials. This paper mainly designs and optimizes novel porous framework Materials by computer molecular simulation technology, then explores the adsorption amount of methane on these Materials, and discusses the mechanism of methane adsorption of metal framework Materials.
In this paper, the preliminary design of metal organic framework Materials organic linkers was firstly carried out through commercial software Materials Studio. Then, 1138 MOF Materials with different metal centers and topologies were obtained using ToBaCCo3.0 software, and then the Forcite module of Materials Studio software was used to optimize the designed MOF Materials. Using the Zeo software, we can calculate the pore size, specific surface area, porosity and other structural properties of the obtained MOF Materialss.50 Materials are randomly selected from 1138 Materials which were simulated by the Grand Canonical Monte Carlo (GCMC) method at a temperature of 298K and a pressure of 5.8MPa and 65MPa. then we construct a multivariate linear regression model (MLR) between adsorption amount and three properties. The model predicts the methane adsorption capacity of 1138 Materials, and then selects the CH4 adsorption delivery capacity from the descending order of working capacity. The optimal 350 Materials were simulated by GCMC method to obtain the methane adsorption delivery capacity, and five best Materials were selected. The adsorption delivery capacity of these five Materials was simulated under the condition of normal temperature 298K and pressure between 1-80 MPa, and the adsorption isotherms of five kinds of porous Materials for methane were obtained. Finally, the mechanism of methane adsorption of the Materials structure can be obtained by the radial distribution function and the density distribution of these Materials.
According to the simulation calculation, the MOF Materials with fcu topology and zirconium metal has the optimal methane adsorption delivery capacity at 298K, between 5.8MPa and 65MPa.The optimal performance Materials is named fcu_(Zr6O8)(TRZ)12_link23. Through the radial distribution curve and the centroid density map of the Materials, it can be found that the methane molecules are preferentially adsorbed on the metal center at 5.8 MPa, and preferentially adsorbed on the organic linker at 65 MPa.
Key words: Adsorption of CH4; Porous framework Materials; Molecular simulation; Quantum mechanics calculation;
目 录
摘 要 I
Abstract II
目 录 1
第一章 绪论 1
1.1. 概述 1
1.2. 选题的意义 3
第二章 理论计算原理与方法 5
2.1. 分子模拟简介 5
2.2. 分子模拟在多孔材料中的应用 6
2.3. 力场模型 6
2.3.1. 力场概述 6
2.3.2. 甲烷和MOF材料的LJ势能参数 7
2.3.3. 交互作用势能参数 7
2.4. 巨正则蒙特卡洛模拟 8
2.5. 径向分布函数 8
2.6. 等量吸附热 9
2.7. 质心密度分布 9
第三章 MOF材料建模与初步筛选 10
3.1. 有机连接体设计 10
3.2. 分子模型建立 10
3.3. MOFs模型优化及结构参数计算 12
3.4. 甲烷存贮量-结构关系与初步筛选 12
第四章 结果与讨论 14
4.1. 最优甲烷存贮性能材料 14
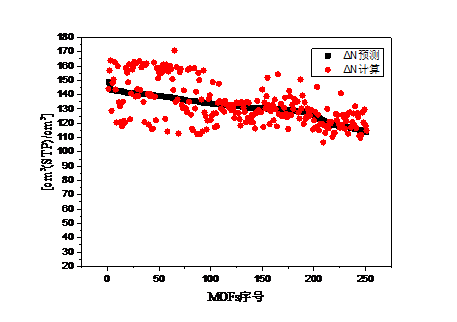
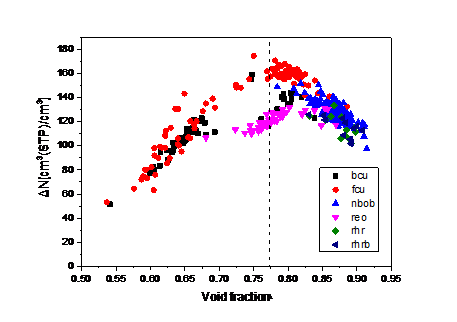
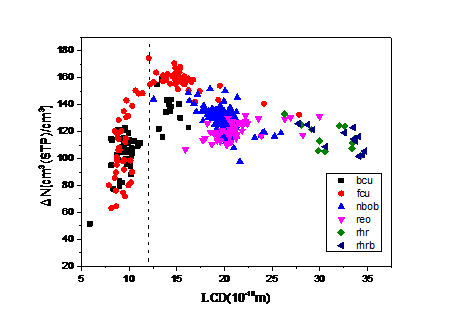
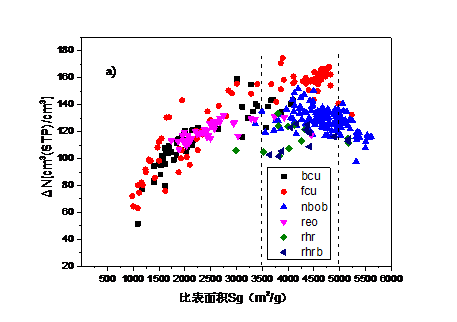
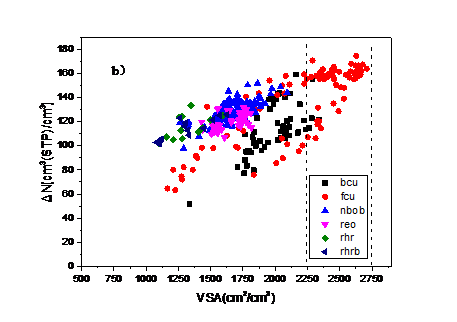
4.2. 吸附最大工作容量与结构参数和吸附热的关系 15
4.3. 甲烷在五种材料中的吸附等温线 17
4.4. 材料CH4存贮机理的分析 18
4.4.1. 材料fcu_(Zr6O8)(TRZ)12_link23的径向分布函数 18
4.4.2. 最优存贮性能材料的构型图与质心密度图 19
第五章 结论与展望 21
参考文献 22
绪论
概述
由于化石燃料的燃烧,大气中二氧化碳含量的持续增加是当今最为关键的全球环境问题之一,并且正在推动对替代燃料的强烈追求。天然气(Natural Gas, NG)被认为是一种对环境危害较小的能源。天然气的主要组成是甲烷,被认为是一种有前景的“桥梁”燃料:一种有助于向无碳燃料过渡的中期解决方案。特别在当前,人们对开发天然气燃料汽车产生了相当大的兴趣。天然气已经用于发热与发电,但尚未广泛应用于运输,尽管燃烧时产生的二氧化碳比传统汽油或柴油少25%,但甲烷相对较差的体积能量密度限制了天然气燃料车辆的广泛商业化。目前,天然气储存主要有液化天然气(LNG,111 K)、压缩天然气(CNG,25 MPa)、吸附天然气(ANG)三种[1]。LNG需要在低温下储存,存在液化天然气瓶维修保养困难的问题,同时燃料站建设投资巨大,经济性差,对于车用而言这种储气方式的实用性有限。CNG是通过压缩将天然气储存在高压容器内,是目前车载主要的储气方法,但也存在一定的缺陷,例如高压设备的制造工艺复杂且成本过高、高压设备自身的安全隐患。因此在这种情况下,人们提出吸附天然气作为替代方案,ANG是指在容器中装入具备微孔结构和高比表面积的专用吸附剂,可在常温、中低压下将NG吸附储存的技术,用范德华力替代储存所需要的部分压力使NG分子吸附于吸附剂微孔内表面,增加了天然气的储存密度,克服了压缩天然气的不足,因而具有广阔的应用前景[2]。ANG技术的重点是选择和设计制备性能优异的吸附剂。
影响ANG吸附剂的吸附性能的因素有:(1)吸附剂是否具有适宜的微孔结构和较大的比表面积;(2)吸附剂是否具备高效的天然气储存能力和尽可能大的单位体积吸附量;(3)吸附剂是否具备良好的导热性能;(4)正常情况下,吸附剂是否具有较快的吸脱附速率;(5)吸附剂是否具备较长的使用寿命和可再生性;(6)制备工艺是否简单且成本较低[3]。自19世纪50年代起,人们就开始筛选各种适合于甲烷储存用的吸附材料,如天然沸石、分子筛、活性氧化铝、硅胶、碳黑、活性炭等[3]。多种多孔材料是天然气储存的潜在候选材料,如多孔芳烃骨架(PAFs)、共价有机骨架(COFs)、多孔聚合物网络(PPNs)和金属有机骨架(MOFs)[4]。这些新型多孔材料大多具有比表面积高、孔隙率高、结构可设计性好及良好的热稳定性良好等优点,在气体储存、气体分离和分子催化等领域具有广阔的应用前景。
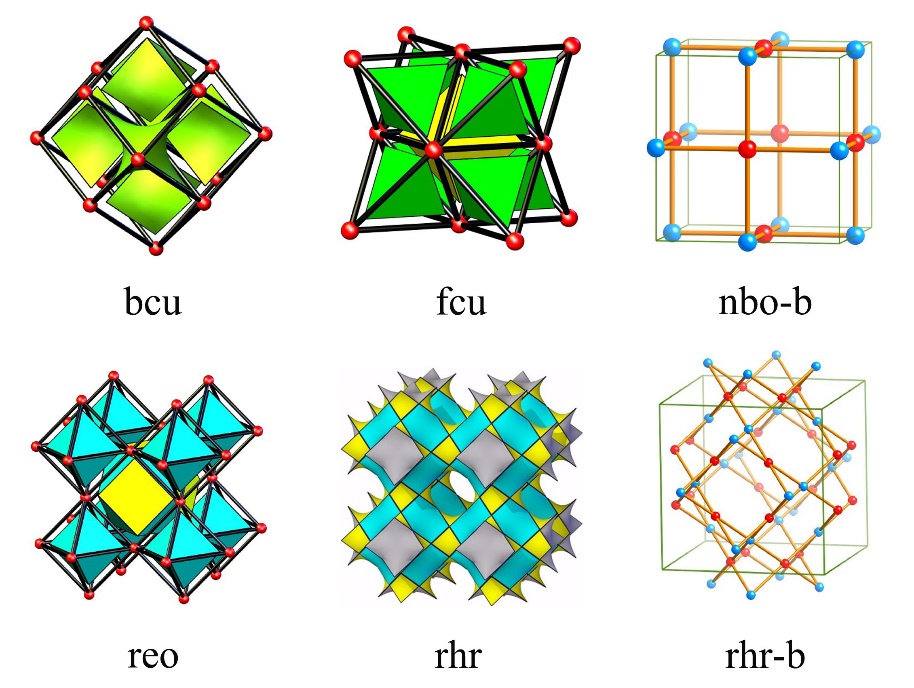
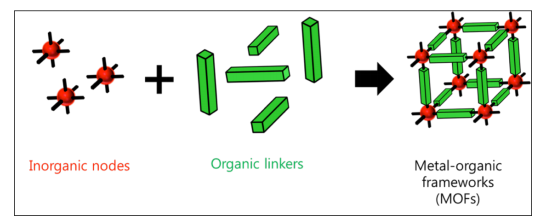 金属有机骨架(MOFs)是一类相对较新的纳米多孔结晶材料,由无机金属中心节点(Inorganic nodes)和有机连接体(Organic linkers)自组装构成(如图1.1),因为有大量可能的构建块,所以存在几乎无限数量的潜在MOFs[5]。这些材料具有几个重要的性能,包括超高表面积,低骨架密度,明确的多孔结构,可调的化学功能,以及良好的热和机械性能,这使它们成为一种有前途的多孔材料,其应用范围非常广泛,如气体储存,分子分离,化学传感,催化,药物输送等。特别是在气体储存和分离领域,已经发现MOFs比传统的吸附材料如沸石和活性炭具有一些重要的优势。
金属有机骨架(MOFs)是一类相对较新的纳米多孔结晶材料,由无机金属中心节点(Inorganic nodes)和有机连接体(Organic linkers)自组装构成(如图1.1),因为有大量可能的构建块,所以存在几乎无限数量的潜在MOFs[5]。这些材料具有几个重要的性能,包括超高表面积,低骨架密度,明确的多孔结构,可调的化学功能,以及良好的热和机械性能,这使它们成为一种有前途的多孔材料,其应用范围非常广泛,如气体储存,分子分离,化学传感,催化,药物输送等。特别是在气体储存和分离领域,已经发现MOFs比传统的吸附材料如沸石和活性炭具有一些重要的优势。
图1.1 示意图展示了MOFs如何从金属中心节点和有机连接体自组装形成多孔框架
早在1999年,加州洛杉矶大学的Yaghi团队合成出了MOF材料[6](又称MOF-5),这是最早报道的具有稳定多孔结构的MOF材料,由于其具有较大的比表面积和自由孔体积,因而表现出极佳的气体吸附性能。之后,该课题组又合成出以Zn4O(CO2)6为固定金属中心的一系列IRMOF-n材料,并对这一系列材料做了不同气体吸附的测试,发现甲烷吸附性能优良,其中IRMOF-1和IRMOF-3在29K下甲烷存贮量分别到达135和120cm3(STP)/cm3, 特别的IRMOF-6材料吸附量到达155 cm3(STP)/cm3,这一数值远高于传统的多孔材料[7]。除此之外还有一大批实验合成与计算模拟的具有很高的甲烷存贮能力的MOF材料已被报道,例如UTSA-76a[8]材料在5.8MPa到65MPa间的甲烷存贮能力已达到187 cm3(STP)/cm3,这是目前所有报道中具有最高的甲烷存贮性能的材料;在同样的工作压力下,HKUST-1[9]和NU-125[9]材料的存贮能力分别为184和174 cm3(STP)/cm3。
随着电子计算机模拟技术的快速发展,在不同的学科领域分子模拟技术都有着非常广泛的应用。对于化学化工和材料领域来说,不同于传统上的实验方法,计算机分子模拟主要是通过建立模型,从更加微观的层面对相关问题做讨论,然后将问题微观的性质通过叠加方法转换成宏观性质。分子模拟方法对实验现象进行的模拟和预测主要是针对微观层面,能够起到对实验相关研究方向的指导作用。目前为止计算机化学已广泛应用到金属有机骨架材料研究领域,通过计算模拟可以进行大规模的材料筛选工作,这不仅使得金属有机骨架材料的结构性质的研究速度得到提升,同时也促进了它们的实际应用。
选题的意义
最近很多年来,由于煤和石油等能源的消耗,人类所面临的问题不只是资源短缺,还要面对由于燃烧化石能源产生的污染问题。天然气由于其储量大,产物污染小的优点因此被认为是具有广阔的发展前景的能源。但由于其主要成分甲烷的体积能量密度相对较差,对天然气的运输和储存造成极大的困难,限制了车载天然气的广泛应用。近十几年来出现的金属有机骨架材料由于比表面积大、孔隙率适宜、物理化学性质稳定等优点,在储存气体上具有优异的表现。根据金属有机骨架材料独特的结构特征使得它不仅仅能够用于储存气体,而且在许多别的领域都具有异常广阔的前景。
美国能源部设定的常温下5.8MPa到65MPa间的甲烷体积存贮能力达到315 cm3(STP)/cm3,但是目前最优甲烷存贮性能的材料只达到目标的65%[10],因此找到一种或数种性能更优的材料迫在眉睫。除此之外,通过计算机模拟筛选大量不同类型的MOF材料,结果表明甲烷在MOFs中的存贮可能存在一个极限[11],即存贮能力很难超过200 cm3(STP)/cm3,这就使得进一步探究甲烷在MOFs中的吸附机理变得十分重要。通过课题组前期所发表的文献的研究发现含四唑基的MOF材料表现出良好的甲烷吸附性能,这对于MOF材料的甲烷存贮性能的进一步提高具有重大意义。但是MOF材料的结构多样性在一定程度上增加了实验合成难度。此外,如果使用传统的实验方法研究这个问题,不仅过程繁琐,时间消耗很长,而且没办法对多种多样的金属有机骨架材料进行系统性地研究分析。
现如今计算模拟化学在当今的科学研究中的使用变得越来越普遍,在化学、化工、材料等科学领域都具有十分广泛的应用。计算化学,究其本质就是根据现有的科学理论知识使用十分先进的科学技术,计算模拟体系中原子或分子的物理、化学变化,根据计算模拟得到的结论来预测出相关材料的实际物理化学性质。计算机分子模拟方法具有的优点不仅包括能够缩短实验周期,还可以减少完全不必要的材料合成所导致的浪费资源,还包括能够提供理论指导实际合成材料。本文通过使用计算化学模拟的相关方法,使用密度泛函理论和蒙特卡洛模拟的计算原理,采用GCMC模拟研究了不同金属有机骨架结构材料在常温下及压力5.8MPa-65MPa间对甲烷的吸附的现象。通过使用Materials Studio、Zeo 、ToBaCCo3.0、RASPA等软件对金属有机骨架材料的结构与性质进行设计与计算,从而获得材料的比表面积、孔隙率和孔径等结构参数,并且使用径向分布函数和质心密度分布等理论方法研究甲烷分子周围不同原子的分布情况,探究金属有机骨架材料的甲烷存贮机理,获得甲烷吸附性能与材料结构参数的关系,为设计新型MOF材料提供理论指导,从而可以减少实验合成所造成的不必要的资源浪费,进一步可以解决社会对于车载甲烷存贮的实际应用问题。
理论计算原理与方法
分子模拟简介
分子模拟的定义是使用计算机技术对实际存在分子进行模拟,其优势在于能够通过计算机完善和补充实验的缺陷与不足,通过相关计算可以得到很多无法从实验得到的结果与数据。分子模拟在物理和化学领域都有着很大的应用,人们可以设计某些计算机程序来完成一些物理及化学实验过程,在整个过程中,分子模拟可以观察物理现象、探究化学反应机理,模拟新的化学反应路径。
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: