开放金属铜位点MOFs材料选择性吸附分离烃类混合物的研究毕业论文
2020-02-19 11:52:20
摘 要
金属有机框架材料(MOFs)作为一种新兴多孔材料,具有高孔隙率,结构多样性和可调性等特点,目前在气体储存和气体分离等方面已展现出了巨大的应用前景。大量的MOFs材料给实验研究带来了一定的挑战,采用分子模拟技术对MOFs材料进行计算筛选可快速获取用于目标气体分离的最佳材料,有助于指导实验的正确进行。因此本文主要通过采用巨正则蒙特卡洛(GCMC)模拟方法模拟C2H6/C2H4分子在具有开放性金属铜位点的MOFs中的吸附平衡状态,通过探究 MOFs的结构-吸附性能间关系,筛选出性能最佳的MOFs材料,从而为获得高效选择分离烃类混合物的吸附剂奠定理论与实践基础。
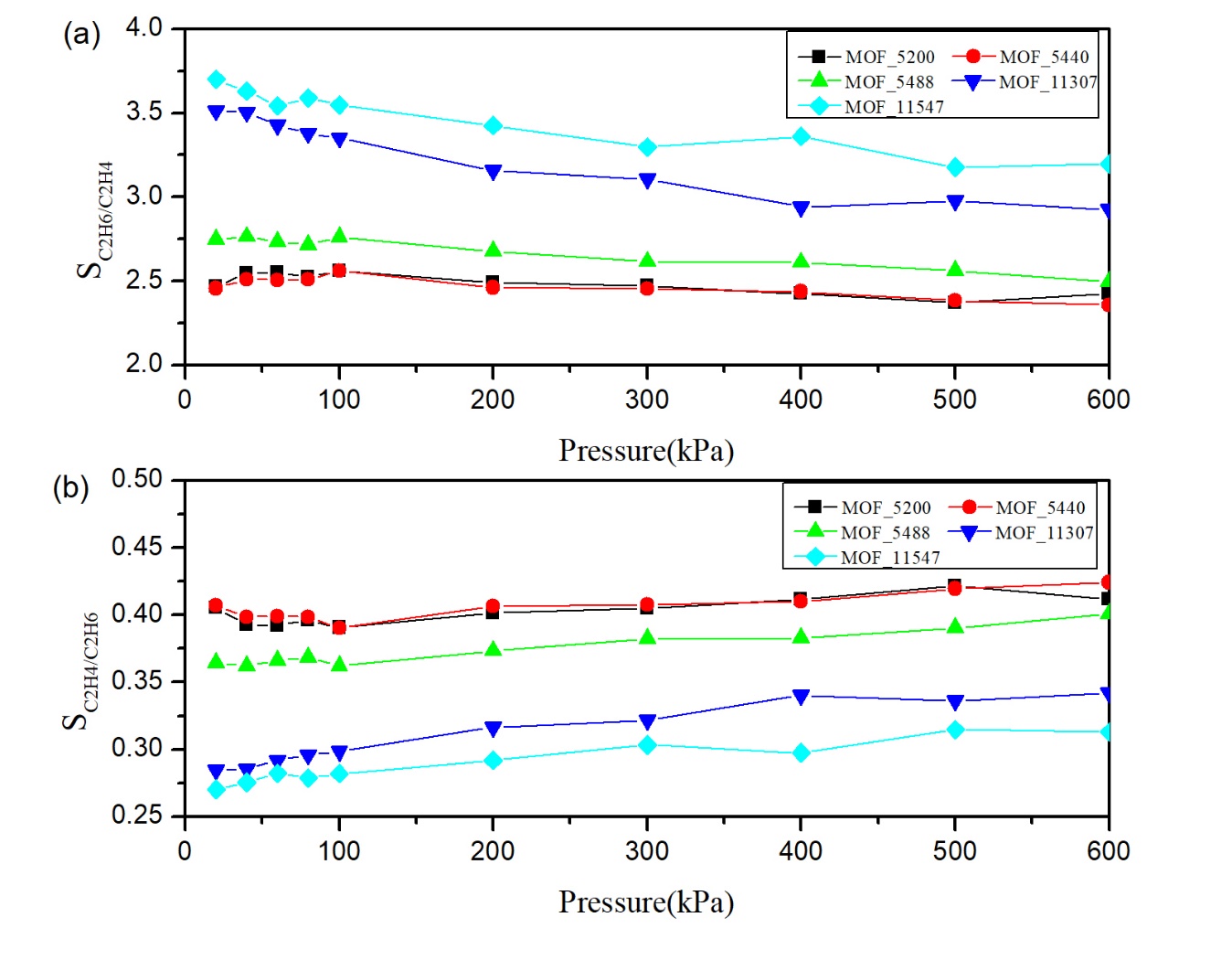
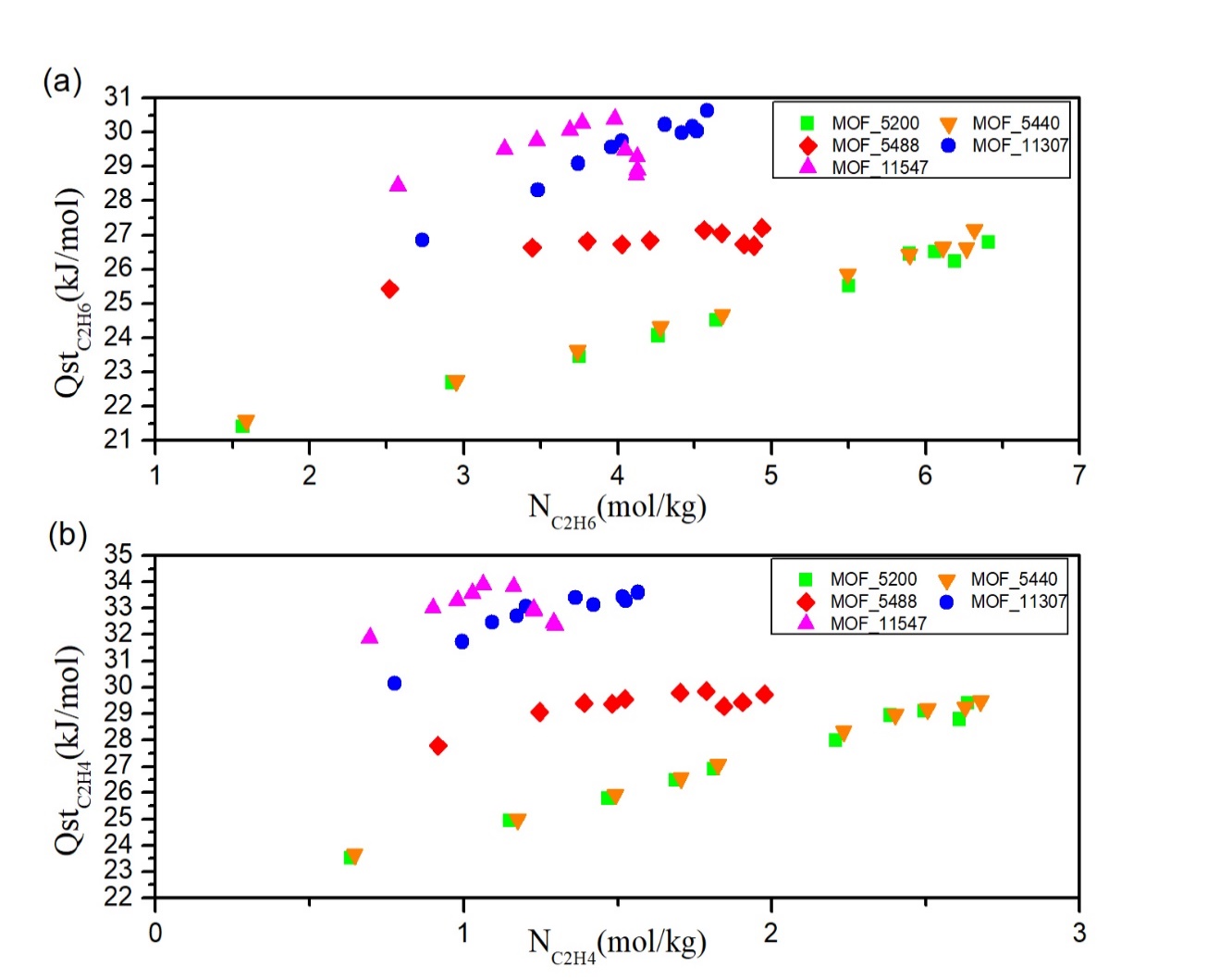
本文基于TOBACCO MOFs数据库中筛选出的5732种具有开放金属铜位点的MOFs材料进行大规模计算筛选。首先利用C2H6/C2H4的动力学直径和MOFs的结构参数筛选出最大孔腔直径(LCD)在4.4-11 Å间MOFs材料共321个。其次,用GCMC模拟298 K,100 kPa条件下C2H6/C2H4混合物在321个MOFs中的吸附量,计算吸附选择性,吸附剂性能评价指数(APS),探究结构-吸附性能间关系。基于APS筛选出5种性能最佳的MOFs材料,模拟计算在298 K,10个不同的压力下5种材料的吸附等温线。最后,通过吸附热、径向分布曲线等数据探究吸附机理。结果表明,MOF-11307相比于其他MOFs材料其分离能力较高,APS为13.3,吸附量为4.03,选择性为3.31。
关键词:金属有机框架材料;烃类混合物;吸附分离;分子模拟。
Abstract
As an emerging type of porous materials, metal-organic frameworks (MOFs) have shown great potentials in the wide application such as gas storage and gas separation, owing to their high porosity, structural diversity and tunability. A large number of MOFs materials have brought some challenges to experimental research, but using molecular simulation technology to calculate and screen the MOFs materials can quickly obtain the best materials for the separation of target gases, which is helpful to guide the correct experiment. Therefore, Grand Canonical Monte Carlo (GCMC) was adopted to calculate the adsorption equilibrium state of C2H6/C2H4 mixtures in MOFs with open metal copper sites in this paper, by exploring the relationship between structure-adsorption properties of MOFs to find the best separation performance materials, which provide a theoretical and practical basis for obtaining efficient selection of adsorbent for the separation of hydrocarbon mixtures.
In this paper, basing on the 5732 MOFs materials with open metal copper sites that screened in TOBACCO MOFs database, a large-scale computational screening were performed. Firstly, 321 MOFs with a largest cavity diameter (LCD) of 4.4-11 Å were screened by using the dynamic diameter of C2H6/C2H4 and the structural parameters of MOFs. Secondly, using GCMC to simulate the adsorption capacity of C2H6/C2H4 mixtures in 321 MOFs under the condition of 298 K, 100 kPa, at the same time, the adsorption selectivity, the adsorbent performance evaluation index (APS) were calculated, and the relationship between structural and adsorption properties was explored. Based on APS, screening 5 MOFs with the best performance and simulate the adsorption isotherms of 5 MOFs under 298 K and 10 different pressures. Finally, the adsorption mechanism is explored by adsorption heat, radial distribution curve and other datas. The results showed that the separation ability of MOF-11307 compared with other MOFs materials was higher, the APS was 13.3, the adsorption capacity was 4.03 and the selectivity was 3.31.
Key words: metal-organic frameworks; hydrocarbon mixtures; adsorption separation; molecular simulation.
目录
摘要 II
Abstract II
第1章 绪论 2
1.1 概述 2
1.2 金属有机框架材料(MOFs) 2
1.2.1 MOFs材料结构特性 2
1.2.2 MOFs材料在轻烃分离方面的应用 2
1.3 计算方法在MOFs分离中的应用 2
1.4 选题的依据及意义 2
第2章 模拟细节 2
2.1 MOFs材料筛选 2
2.2 分子模拟 2
2.2.1 巨正则蒙特卡洛模拟计算方法(GCMC) 2
2.2.2 势能模型和力场参数 2
2.2.3 C2H6/C2H4势能模型 2
2.3 MOFs性能评估指标 2
第3章 结果与讨论 2
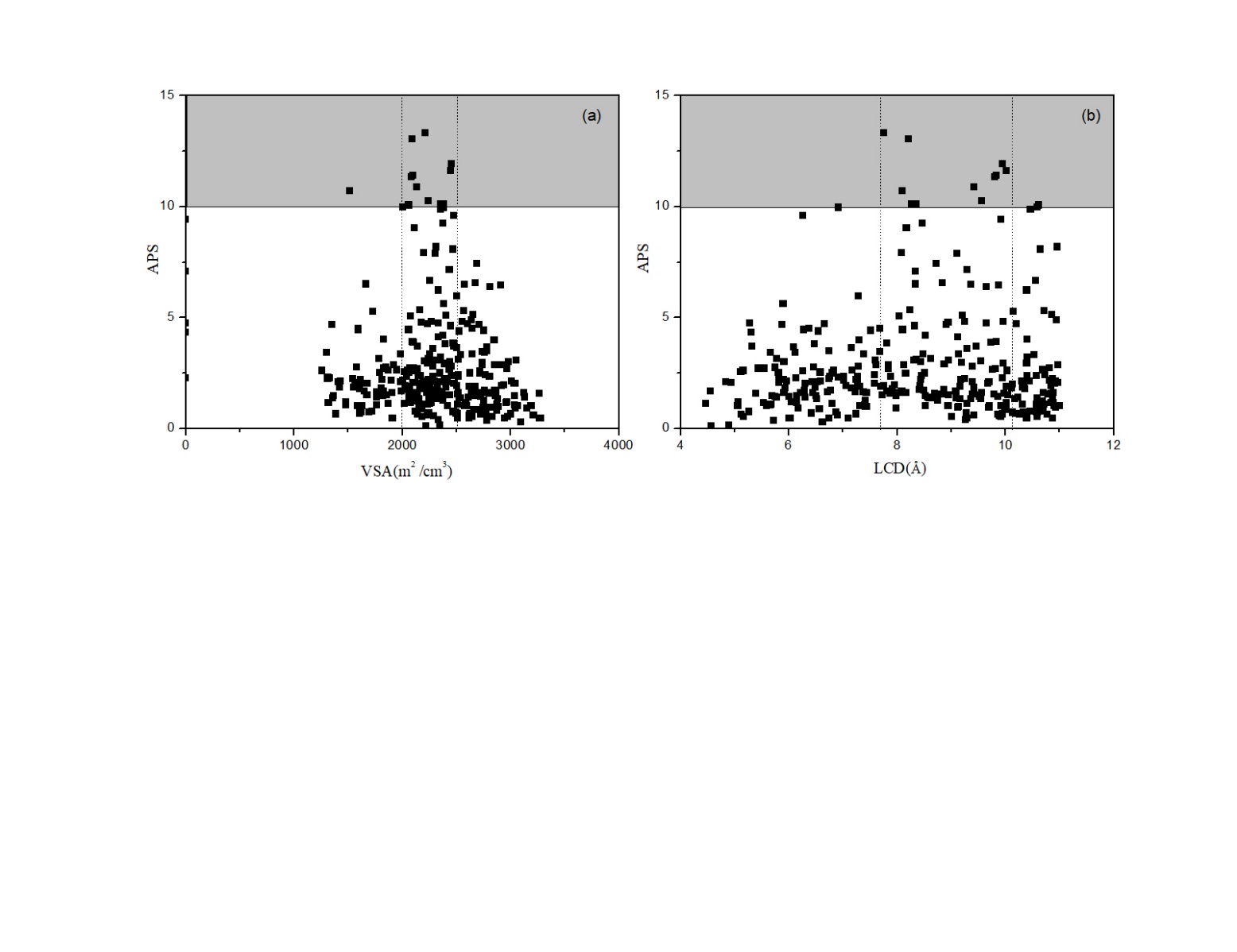
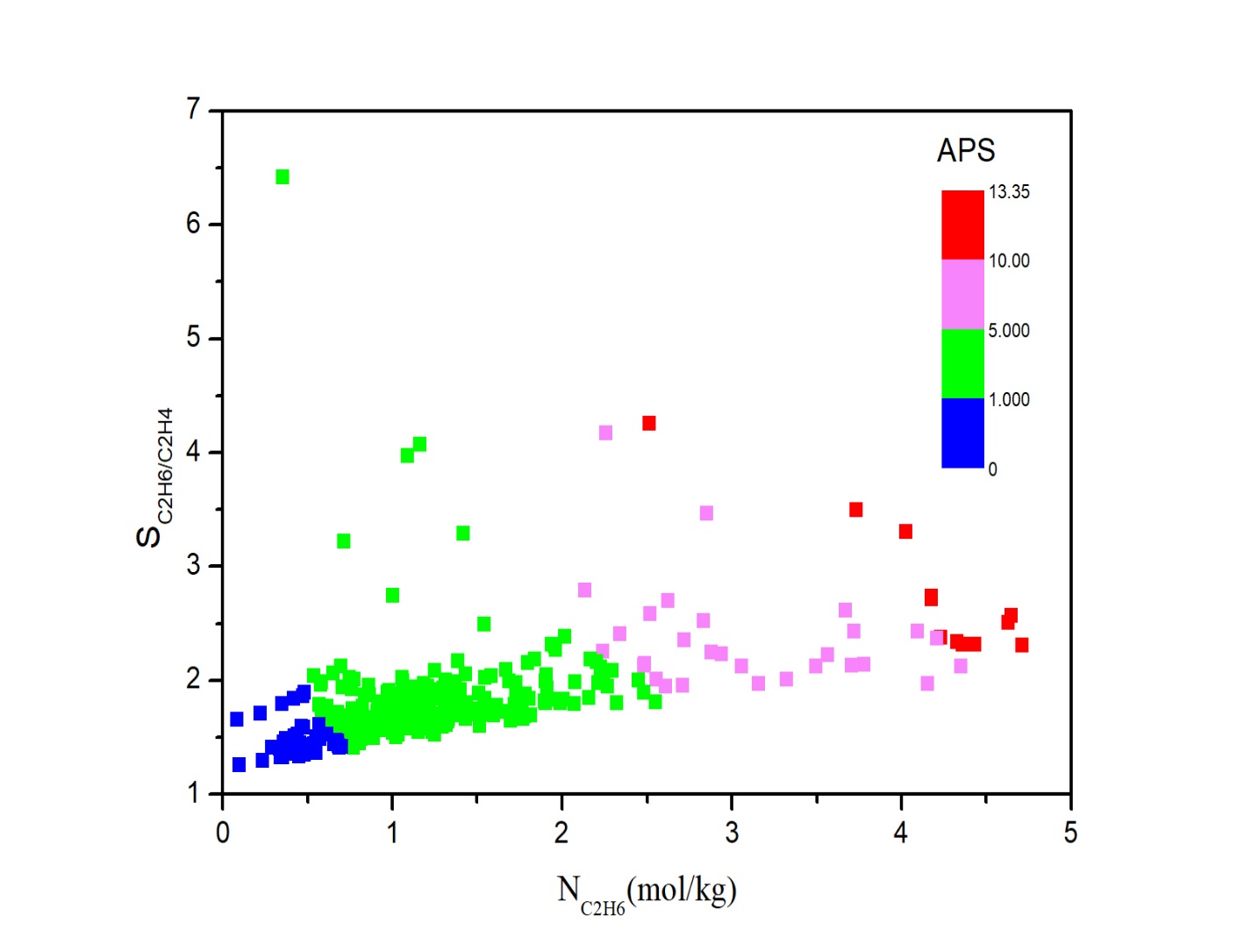
3.1 MOFs分离性能 2
3.1.1 结构-性能关系 2
3.1.2 MOFs材料排序 2
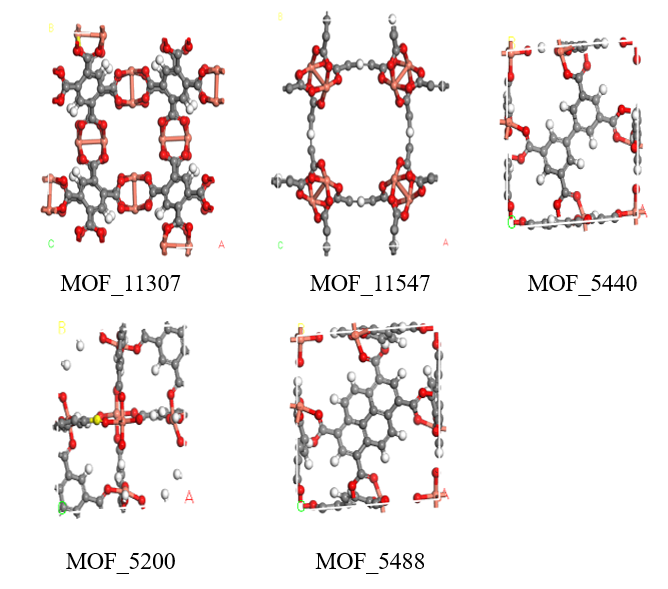
3.2 五种TOP-MOFs 2
3.2.1 优选材料的分子模型 2
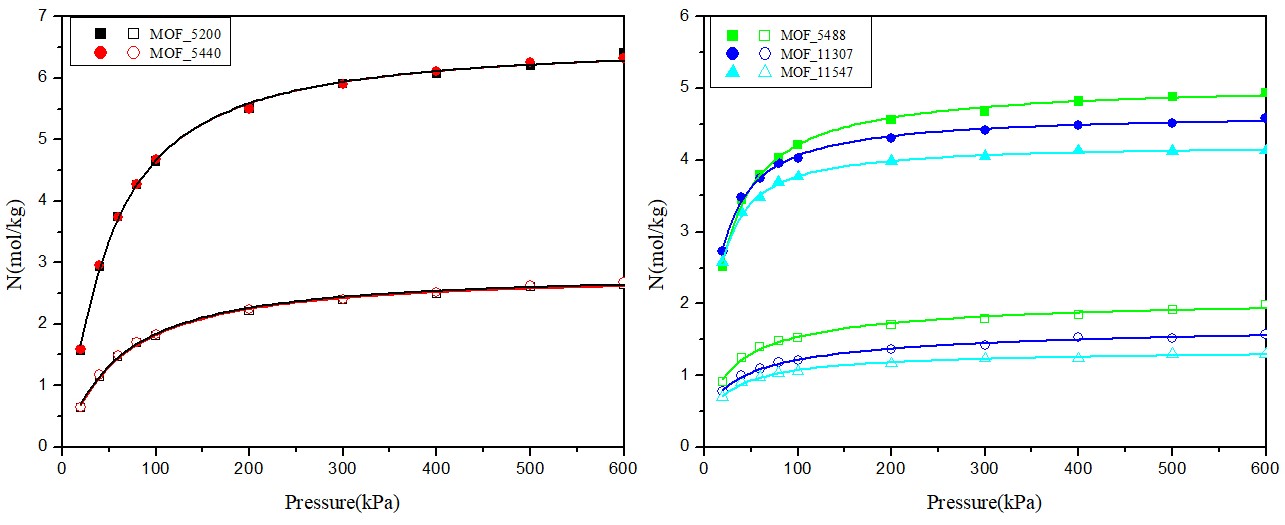
3.2.2 吸附等温线 2
3.3 吸附机理分析 2
3.3.1 吸附热 2
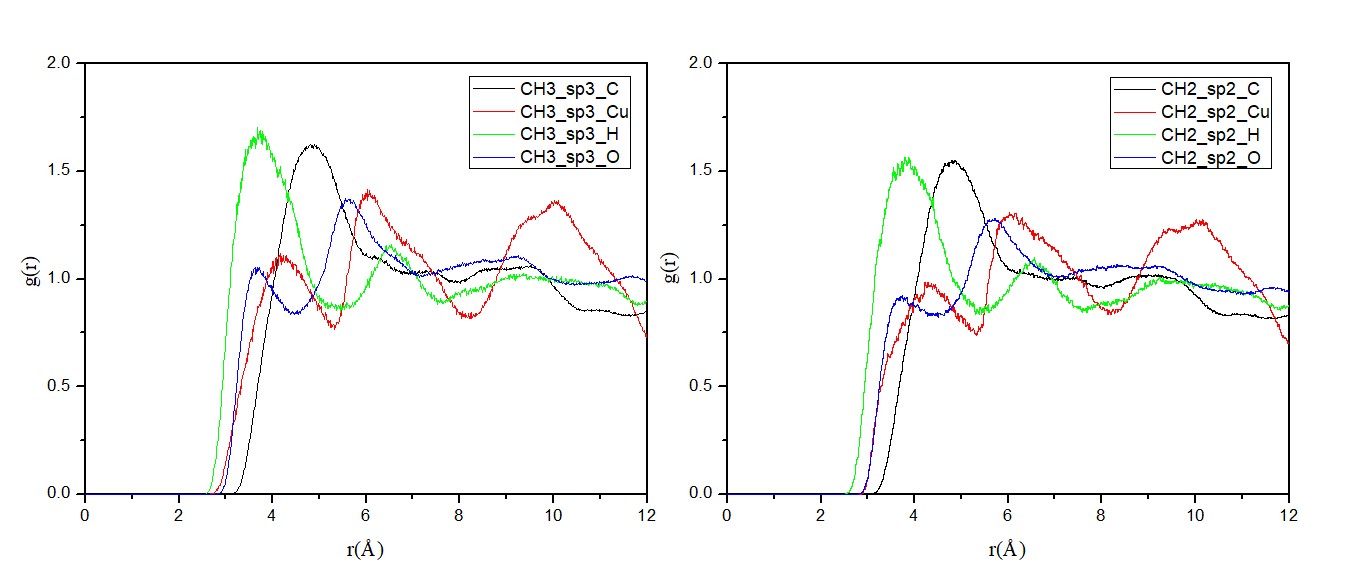
3.3.2 径向分布函数 2
第4章 结论与展望 2
4.1 结论 2
4.2 展望 2
参考文献 2
致 谢 2
第1章 绪论
概述
C2轻质烃,包括乙炔、乙烯和乙烷等,是重要的能源和化学工业原材料。乙烯可用于制造各种石油化工产品,如聚乙烯、聚氯乙烯、乙二醇等[1]。利用乙烯制造的产品可广泛应用于工业、农业、生态等各个领域,其产品占据化工产业总产品的75%以上,乙烯的产量更是衡量一个国家石油化工产业发展情况的重要指标。因此,乙烯的分离和纯化是石油化工产业的重要工艺过程,工业上乙烯的来源主要通过石油裂解的方法,裂解气所含的主要杂质为乙烷。由于乙烯和乙烷分子沸点相近,结构相似,且相对挥发度较小,采用传统低温精馏工艺分离乙烯/乙烷存在能耗大、成本高、分离效果差等缺点,因此工业上迫切需要发展一种新型分离技术来替代传统工艺。目前,随着分子筛,活性炭、多孔氧化铝等吸附分离材料的发展,吸附分离技术在气体分离方面展现出了巨大的应用前景[2]。和传统低温精馏工艺相比,其具有能耗低,操作简单,吸附选择性较高等优点[3, 4]。
吸附分离技术的关键点是在于合适吸附剂的选取。目前吸附剂的种类包括活性炭、沸石分子筛、硅胶等传统吸附剂和金属有机框架材料(MOFs)等新型吸附剂,和传统吸附剂相比,MOFs材料具有比表面积大、孔隙率高、可功能化修饰、结构多样化等优点,在气体吸附分离方面展现出了较高的吸附量和选择性,已成为具有广泛应用前景的吸附剂候选材料。
近年来,MOFs材料在选择性吸附分离乙烯/乙烷等烃类混合物的研究方面已取得多项成果。如Bao[5]课题组将Ag(I)负载到磺酸化的MOFs孔道内,研究303 K时,等量的乙烯/乙烷混合物在MOFs材料Cr-MIL-101-SO3H中的吸附分离过程,研究结果表明该材料对乙烯的吸附能力较强,分离选择性可达16。目前,MOFs材料的数量正在迅速增加,其种类多样,数量巨大,利用实验方法对MOFs材料的分离性能进行逐一测试显然是不切实际的。随着科技的发展,计算机分子模拟技术已成为研究吸附分离行为的重要辅助手段,其主要通过建立分子模型来模拟分子的结构与行为,利用分子模拟技术可对MOFs材料进行快速筛选从而获得具有优异分离性能的目标材料。
金属有机框架材料(MOFs)
MOFs材料结构特性
金属有机框架材料(MOFs)是一种由金属离子或金属簇与桥联的有机配体自由组装而形成的新型有机-无机杂化材料[6]。其作为一种跨越有机无机化学、材料化学、晶体拓扑学、配位化学、超分子化学等多个学科的新型多孔材料[7],近年来引起了研究者们的广泛关注。目前,MOFs材料已在气体储存分离[8, 9]、催化[10]、分子识别[11]等领域得以广泛应用,被认为是传统多孔材料的有效替代品,其具有一些传统多孔材料所不具有的结构特性,例如:
- 结构功能多样性:MOFs材料金属中心的种类涉及到主族金属、过渡金属以及镧系金属等,同一金属的多种价态也会导致MOFs材料的结构有所不同。MOFs材料的有机配体既可以是有机酸、有机碱,也可以是磷酸酯、磺酸酯、胺类、吡啶类等配体。金属中心和配体的多样化使得MOFs材料结构可调,功能多样。
- 高比表面积、高孔隙率:MOFs材料具有高比表面积(达6000 m2/g)、高孔体积(1-4 cm3/g)、多种孔径( 1-98 Å)[12]以及高孔隙率(达0.9),是具有优异物理性质的晶体结构。
- 结构稳定性:MOFs材料已有近30多年的发展历程,早期MOFs材料的骨架结构通常伴随着客体分子的移除而瓦解崩塌,随着对MOFs材料的逐步深入研究,目前,MOFs材料骨架结构在高温、高真空、客体分子移除的条件下仍能保持稳定,柔性MOFs材料的骨架结构更会随着客体分子的种类,外界条件的改变而改变。
MOFs材料在轻烃分离方面的应用
低碳烃是化学工业的重要基本原料和产品,烃类混合物的分离更是化工生产过程的重要工艺之一。近年来,烯烃/烷烃的分离一直是吸附领域研究的热门课题,MOFs材料作为一种新型吸附剂,其可通过利用氢键、配位键等识别机制,或通过精确控制材料的孔径、孔型等多种方法来实现烃类混合物的分离。目前,MOFs材料在烃类混合物的分离研究上已取得了一定的进展。MOF-74系列是一类具有开放性金属位点的MOFs,Bao课题组[13]早期首次报道了采用Mg-MOF-74材料分离烯烃和烷烃,分别测定了在278、298和318 K,压力为100 kPa下C2H4/C2H6,C3H6/C3H8 在Mg-MOF-74吸附剂上的吸附平衡,结果表明烯烃优先于烷烃吸附,C2H4/C2H6,C3H6/C3H8吸附选择性分别为15,19。Snurr等[14]随后研究了MOF-74系列(Mg,Co,Mn)材料对C3H6/C3H8的分离性能,发现相比于其他两种材料,CoMOF-74分离性能最好。Hu等[15]制备了一种微孔MOFs(UTSA100),用于从含有1%C2H2的C2H4/C2H2混合物中除去C2H2。结果显示在296 K和1 atm下,C2H4、C2H2吸收量分别为95.6和37.2 cm3g-1,选择性为10.72。在C2H6/C2H4混合物吸附分离方面,Li课题组[16]合成了Fe2(O2)(dobdc),研究了材料与C2H6的结合性能,测试了C2H4/C2H6混合物的分离性能,发现在较宽的压力范围内,C2H4/C2H6分离选择性较高,二者分离选择性可达4.4。Guo课题组[1]研究了298 K,100 kPa下钙基MOFs材料对C2的吸附分离效果,结果表明C2H2 / C2H4混合物(50:50,v/v)的吸附选择性为8.1,高于常用MOFs材料UTSA-67a,MOF-74系列等,这是因为C2H2可优先与MOFs材料形成氢键和π-π相互作用,而C2H4 / C4H6混合物(50:50,v/v)吸附选择性也可增加到5.9。
计算方法在MOFs分离中的应用
MOFs材料种类繁多,利用实验方法来确定所有MOFs材料的分离性能对实验者来说是一项巨大的挑战,因此,众多研究者选择采用高通量计算筛选方法来筛选出符合目标分离要求的最佳性能的MOFs材料[17]。Jiang课题组[18]利用高通量计算方法筛选了4764个CoRE-MOF用于从烟气(CO2/N2)和天然气(CO2/CH4)中分离出CO2,首次建立了金属类型和吸附剂评估指标间的关系,最终确定了30个最好的CoRE-MOF,发现这些材料大多含有镧系元素。Snurr等[19]针对甲烷存储性能,首次对137953种 MOFs材料采用高通量计算筛选方法,从中发现了300余种高性能存储甲烷的MOFs材料结构。Qiao等[20]利用高通量计算方法筛选出6013种MOFs材料用于从天然气中分离出H2S和CO2,确立了MOFs材料结构与性能间的关系,从中发现了性能优异的MOFs材料。Cao课题组[21]利用巨正则蒙特卡洛模拟(GCMC)方法对23种不同的具有代表性的金属有机骨架材料(MOFs)在Rn/N2和Rn/O2混合物中的选择性进行了研究,发现了ZIF-12、HKUST-1、IMRO-62和ZIF-11四种优良的可用于捕捉Rn的材料。
高通量计算筛选是实验室筛选MOFs 材料的重要手段之一,利用分子模拟技术可从微观上计算出材料的结构特性,模拟客体分子的静态结构和动态行为(如吸附/脱附等),同时可深入的探讨分子间作用机理,深层次的预测尚未合成材料的理化性质,为新材料的研究和开发提供理论基础。相比于实验化学,该方法依靠着强大的计算机计算能力可对化学问题进行精确计算,极大的减小了实验复杂度,降低了实验时间和工作量,节约了实验成本[22]。目前,该方法已在MOFs材料储氢、储甲烷、CO2、烯烃/烷烃的捕集或分离等领域得到广泛应用。
选题的依据及意义
低碳烃是重要的工业原料和产品,传统低碳烃的分离方法主要为低温精馏工艺,该工艺具有高能耗、高成本、分离效果差等缺点。当前,吸附分离技术正凭借其低成本、易操作等优势逐渐被人们所重视,吸附分离技术的关键之处则在于选取合适的吸附剂,MOFs材料作为近年来发展的新型吸附剂材料,具有比表面积大、孔隙率高、稳定性好、结构功能多样化等优点,在烃类混合物吸附分离方向上展现出了优良的性能,但种类繁多的MOFs材料也增加了吸附剂的筛选难度。当前,在C2H6/C2H4混合物吸附分离方向上,大量的MOFs材料仍存在吸附量小、分离选择性差等问题,且对于MOFs材料的筛选方法、材料结构与吸附性能间的关系以及吸附机理等内容的报道目前尚十分有限。因此,本课题拟在课题组的前期研究基础之上,以从TOBACCO MOFs数据库中筛选出的5732种具有开放性金属铜位点的MOFs材料为研究对象,以C2H6/C2H4混合物的选择性分离为应用目标,主要通过巨正则蒙特卡洛(GCMC)进行吸附模拟计算,旨在探索出不同MOFs材料结构与吸附性能间的关系,同时系统的针对所筛选材料选择性吸附分离C2H6/C2H4混合物的机理进行深入分析,尽可能的筛选出吸附量大,吸附性能优良的MOFs材料,为获得高效选择性吸附分离烃类混合物的吸附剂奠定理论与实践基础,同时为进一步研发高性能的MOFs材料提供理论基础。
第2章 模拟细节
MOFs材料筛选
本文首先采用Haldoupis等基于python开发的OMS-detector代码从TOBACCO MOFs数据库(包含13512个MOFs)中筛选出的5732种具有开放性金属铜位点的MOFs材料作为研究对象,利用Zeo 软件计算了MOFs的物理性质,如体比表面积(VSA,m2/cm3)、孔腔极限直径(PLD,Å)、最大孔腔直径(LCD,Å)等。查阅相关文献,发现LCD值大于10 Å以上的MOFs材料对乙烷/乙烯的选择性吸附效果较差,根据乙烯的动力学直径(4.2 Å)和乙烷的动力学直径(4.4 Å),对其中LCD小于4.4 Å的材料进行吸附量模拟计算,结果显示乙烯和乙烷的吸附量值较低,故筛选出LCD值在4.4-11 Å间的MOFs材料,最终得到321种不同的MOFs材料。该321种MOFs材料的VSA值在0-3300 m2/cm3间,PLD和LCD值分别在2.3-11 Å和4.4-11 Å范围内。其中大多数MOFs(296)的比表面积在1256到3000 m2/cm3之间,5种MOFs的比表面积为0 m2/cm3,20种MOFs具有较高的比表面积(gt;3000 m2/cm3)。筛选出的321种MOFs材料的LCD均大于PLD,其中有41种 MOFs 材料PLD和LCD几乎相等。
分子模拟
巨正则蒙特卡洛模拟计算方法(GCMC)
蒙特卡洛模拟算法(Monte Carlo,MC),又称随机抽样或统计试验方法,是一种以概率和统计理论为基础,通过构造随机数从而有效求解数值计算问题的方法[23]。常用的蒙特卡洛模拟方法的系综涵盖宏观正则系综(NVT)、巨正则系综(VTµ)和微正则系综(NVE)等,其中,基于巨正则系综的蒙特卡洛算法(Grand Canonical Monte Carlo,GCMC)是目前研究工作中最常用的分子模拟方法。在巨正则系综中,粒子和能量可进行自由交换,但化学势(µ)、温度(T)和体积(V)保持不变。其计算过程利用概率行走的马尔科夫链产生不同的微观态,通过统计分析得到体系的热力学平衡状态,可模拟真实状态下吸附体系的吸附动态平衡[24]。因此,本文主要采用GCMC来模拟具有开放性金属铜位点的MOFs材料对C2H6/C2H4的吸附平衡状态,模拟细节如下:
- 采用RASPA 模拟代码进行GCMC模拟,在模拟过程中将所有骨架原子都当作固定处理,客体分子视为刚性分子。模拟时假设有一体积为V的晶胞,该晶胞四周聚集了一定数量的粒子,将晶胞沿各个方向进行周期性的复制,从而可利用周期性边界条件来消除边界效应的影响。由于晶胞大小与模拟计算量密切相关,为提高模拟的计算效率,本论文中选取1×1×1的单元晶胞。
- 模拟时选定截断半径。在计算某个原子的能量时,只计算以截断半径为半径的球体范围内其他原子对该原子的作用力,而超过截断半径以外的作用力可忽略不计。由于截断半径的大小直接关系着模拟计算效率和计算结果的准确度,故本次模拟设置分子间截断半径为12 Å,确保晶胞的尺寸超过截断半径的2倍。
- 设置模拟条件:设C2H6/C2H4混合物的体相组成比为1:1,温度为298 K,压力为100 kPa。每个MOFs材料模拟的总循环步数设为20000步,其中前10000步为平衡吸附状态阶段,后10000步为计算系综状态平均值阶段,每隔1000步输出一次吸附计算结果。每个模拟循环均考虑三种不同类型的移动,包括分子的平移,旋转和交换。
- 当压力较高时,会与气体的真实压力产生偏差,此时采用彭-罗宾逊状态方程(Peng-Robinson Equation)将压力转换成相应的逸度。
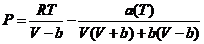 (2.1)
(2.1)
其中:
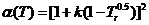
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: