含钌化合物催化乙烯脲氢化还原生成甲醇和乙二胺的反应机理研究毕业论文
2020-04-17 15:07:19
摘 要
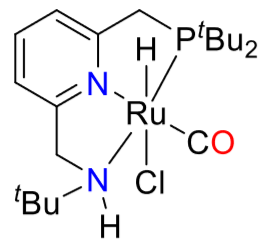
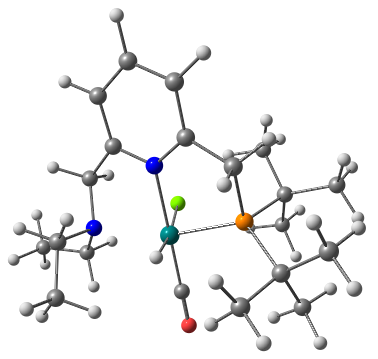
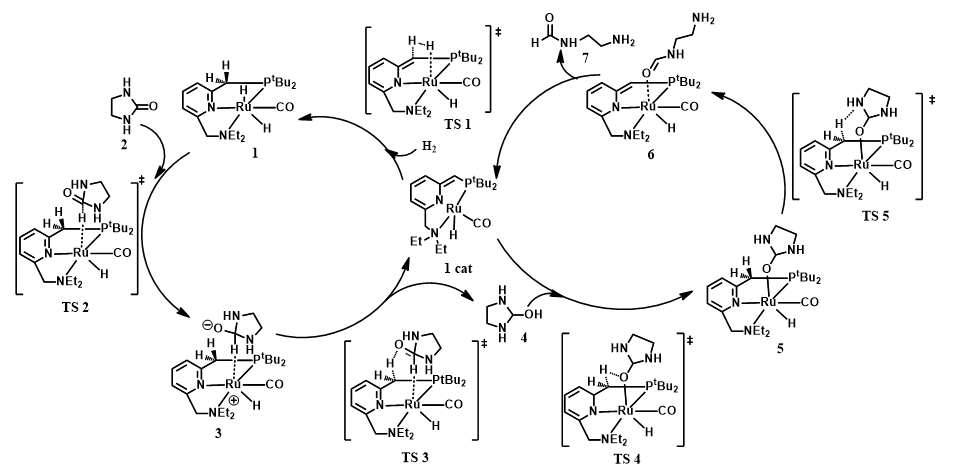
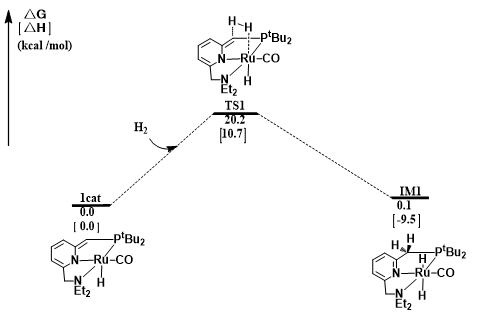
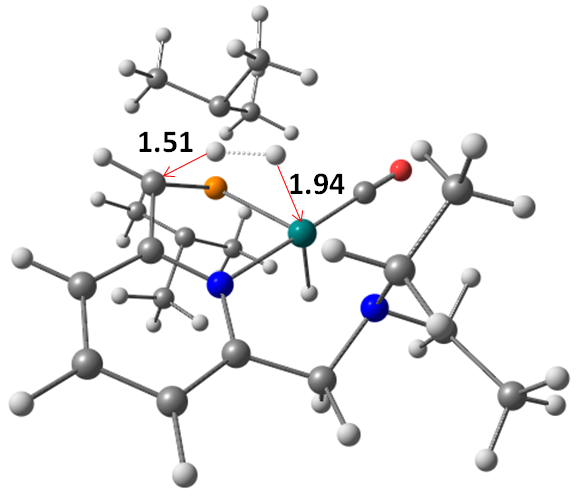
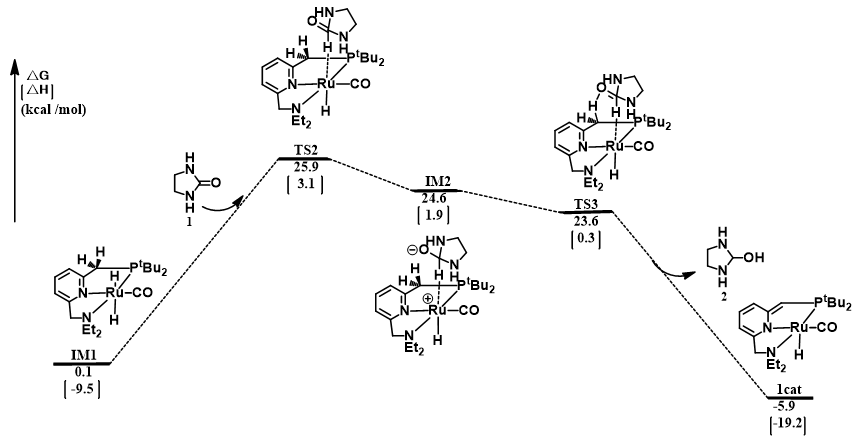
氢能凭借其热值高、燃料利用效率高、无污染、资源丰富等优点被认为是21世纪最有希望的能源之一。几十年以来,人们研究了各种各样的储氢方法,其中液态有机氢载体(LOHC)储氢以其储氢容量高、运输方便安全等特点得到了人们广泛的关注。乙烯脲作为潜在的LOHC储氢体系,其氢化反应机理并不清楚。本论文采用密度泛函理论(DFT)研究了含钌化合物催化乙烯脲氢化还原生成甲醇和乙二胺的反应机理。计算结果表明,整个催化过程主要包括三个阶段:(1)乙烯脲转化为N-(2-氨基乙基)-甲酰胺;(2)N-(2-氨基乙基)-甲酰胺转化为乙二胺和甲醛;(3)甲醛转化为甲醇。此外,乙烯脲的活化能垒最高(25.8 kcal/mol),是整个催化过程的决速步。
关键词: 乙烯脲 甲醇 乙二胺 反应机理 密度泛函理论
Mechanistic study of the hydrogenation of ethylene urea to methanol and ethylenediamine catalyzed by Ru complexAbstract
Hydrogen is considered as one of the most promising energy sources in the 21st century due to its high calorific value, high fuel efficiency, pollution-free and abundant resources. Over the past decades, a variety of hydrogen storage methods have been studied. Among them, liquid organic hydrogen carrier (LOHC) hydrogen storage has attracted wide attention because of its high hydrogen storage capacity and convenient transportation. It is reported that ethylene urea is one of the most promising LOHC systems, however, its hydrogenation mechanism is not clear. Density functional theory (DFT) computations have been carried out to study the mechanism of the hydrogenation of ethylene urea to methanol and ethylenediamine catalyzed by Ru complex. The energetic results show that the catalytic transformation includes three sequential stages consistently involving the catalyst: (1) transformation of ethylene urea to N-(2-aminoethyl)formamide; (2) transformation of N-(2-aminoethyl)formamide to ethylenediamine and formaldehyde; (3) transformation of formaldehyde to methanol. The activation of ethylene urea, which has the highest energy barrier (25.8 kcal/mol), is the rate determining step.
Key Words: ethylene urea ; methanol; ethylenediamine; reaction mechanism; Density Functional Theory
目 录
摘 要 II
Abstract III
第一章 综 述 4
1.1引言 4
1.2储氢技术概述 4
1.3液态有机氢载体(LOHC) 5
1.4量子化学 6
1.4.1计算化学的发展 6
1.4.2量子计算方法 7
1.4.3基组 9
1.4.4过渡态理论 10
1.4.5量子化学计算软件 11
1.5论文研究的意义及内容 12
1.5.1实验意义 12
1.5.1实验内容 12
第二章 实验部分 14
2.1引言 14
2.2实验方案 14
2.3计算方法 14
第三章 实验结果与讨论 15
3.1实验结果 15
3.2未来展望 25
致 谢 28
第一章 综 述
1.1引言
在全球石油天然气资源匮乏的今天,寻找新型的清洁能源正在成为世界各国努力发展的方向。氢能以资源丰富、产热效率高、产物清洁环保等优点被认为是21世纪最有挑战性的能源之一[1]。氢能凭借其清洁、高效、安全、可持续等优点,常常作为燃料应用于航天航空领域。伴随着全球化工材料行业的蓬勃发展,储氢材料更加广泛的被开发和应用于燃料电池方面。因此储氢技术的提升对能源高效利用、材料可持续发展、保护环境等方面都有着巨大的影响。
如何评价一种储氢技术的优劣程度主要凭借以下几个方面:储氢密度、运输成本等。但氢气的特点是密度小、沸点低、难以压缩液化并且高压下易与金属容器形成氢化物从而降低其强度。同时氢气易燃、易爆、易扩散。因此,我们迫切的需要寻找到一种能耗低、储氢密度大、常温常压下可以操作和运输并且可以大范围普及和应用的储氢技术。
1.2储氢技术概述
随着人们对储氢技术的深入研究,储氢技术得到了前所未有的发展。目前,研究和报道最多的储氢技术包括加压储氢[4]、低温液态储氢[5]、合金储氢、活性炭或其他碳材料储氢[6-7]、金属有机骨架材料(MOFs)储氢和液态有机氢载体(LOHC)储氢等。其中,加压储氢的方法研究的最早,应用也最为广泛。但是其储氢质量密度较低,并且加压储氢时会产生氢脆现象,存在潜在的危险性;低温液态储氢虽然体积能量密度高,但是液化过程中所消耗的能量也高,并且需要绝热性能良好的液化罐,对材料要求较为严格;合金储氢的优势在于体积储氢量大,但氢本身会使储氢合金材料变质,如氢损伤、氢腐蚀、氢脆现象等,并且无法实现可循环利用;活性炭或其他碳材料储氢时吸附温度较低(活性炭)或体积储氢量小(碳纳米管),使其应用范围受到限制;MOFs 储氢受制备条件影响较大,目前仍处于研究阶段。
随着科学技术的发展,人们愈加重视氢能源的加工和利用。因此,氢的储存和运输成了亟待解决的问题。传统工艺下的氢储存技术,在价格、安全性、存储效率等各个方面都有不足;与之相比,近些年新出现的LOHC储氢具有以下几个优点:
- 催化过程可逆。LOHC储氢系统反应过程通常都是可逆的、底物可以重复使用、反应产率高。
- 储氢密度高。以下文我们所介绍的乙烯脲储氢循环系统为例,该有机液体储氢载体体系理论上具有6.52wt%的高储氢量,并且在装载和卸载氢气方面具有优异的产率。
- 储氢效率高。例如十氢化萘储氢循环系统,效率可以达到95%。
- 运输方便、安全。运输前夕,LOHC可以生成相应的液态氢化物,到达目的地后,发生脱氢反应重新释放氢气。降低了运输过程中的损失,尤其是在管道运输过程中大大降低了能耗,并且LOHC的维护安全又方便。
- 成本低廉。LOHC储氢系统原则上可以在常温常压下储存和运输,大大降低了保存成本。
1.3液态有机氢载体(LOHC)
随着科学技术的发展,人们也对液态有机氢载体有了更加深入的了解和认识。1975年,由液态芳香族化合物(苯[9-10]、甲苯[11])作为氢载体的液态有机储氢材料由Sultan[8]首次提出。除此之外,大量文献也报道了一些原料易得、氢转化率较高。熔沸点区间合适的LOHC系统,例如顺式-十氢化萘、反式-十氢化萘、环已基苯、咔唑、甲基环己烷等。
Pez[12-15]等最早运用密度泛函理论预测液态有机氢载体。研究中发现,将氮原子引入多环芳香烃中进行取代,可以在很大程度上降低脱氢的反应焓,使得脱氢温度降低。科学家们最早发现,乙基咔唑可以在200℃下实现完全加脱氢,这便给有机物在液态条件下脱氢提供了可能性。在同一阶段,空气化工产品公司进行了杂环芳香有机物在液态有机物储氢技术中的应用,其中提到乙基咔唑中氮原子的引入可以显著降低脱氢反应温度,并且没有产生会引起燃料电池中毒的氨气或一氧化碳等有害产物。这一研究结果使得液态有机物储氢技术应用在汽车动力中成为可能。
上述结果报道之后,引起了储氢材料界的广泛关注,并且推动了一批液态有机物加脱氢的研究和探索。Clot[16]等通过理论计算分析吲哚、喹啉等液态有机物的脱氢性能,提出五元环比六元环更容易脱氢,同时还预测了有利于脱氢反应的有机物的结构。研究中的新型液态有机物储氢剂还包括芘、芴、茚、吲哚、喹啉、咔唑、甲基等芳香杂环化合物。有机液体氢化物储氢是一项很有前景的技术,储氢容量高、运输方便安全,可以实现大规模、长距离、长期性的氢能存储和运输,为车用燃料电池提供氢源[17-18]。目前研究中存在的主要问题为传统有机液体氢化物作为储氢介质,加氢和脱氢温度较高,导致装置费用较高、使用寿命短;贵金属催化剂成本较高,且易中毒失活;非贵金属催化剂成本低,但在使用过程中加氢和脱氢效率较低;若用于燃料电池,加氢时压力较高的问题仍需解决。
1.4量子化学
量子化学是理论化学的一个分支,它是一门将数学、物理和化学学科融合在一起进行化学问题研究的新兴学科。其应用非常广泛,核心就是求解分子的薛定谔方程。薛定谔方程为:
Hψ = Eψ (1-1)
量子化学计算功能十分强大,可以应用于许多不同体系,已经成为了当前化学研究的重要理论工具。
1.4.1计算化学的发展[19-25]
近些年来,计算化学在化学学科的发展过程中,起到了不可忽视的作用,计算化学的发展,根植于量子化学体系而发展。从19世纪末到20世纪初,物理学发生了一场重大的革命,科学开始进入了现代科学发展的时期。量子化学的发展已经经历了100多年,期间,科学家们利用量子力学解释了许多经典物理学理论无法解释的实验现象(如黑体辐射、光电效应、原子光谱和电子衍射等)。随着量子力学的逐步发展,人们意识到用量子力学原理也可以对分子的结构问题进行讨论,因此一个分支学科—量子化学逐渐形成。
20世纪以来,随着Schrödinger、Heisenberg和Dirac等科学家提出并建立了量子力学体系,经过一代代科学家们的研究、钻研,量子化学不再仅是一种理论指导,而在计算领域有了具体的研究方法。这些方法的出现,进一步拓展了量子化学的应用范围,促使了“计算化学”这一独立学科的快速发展。1998年诺贝尔化学奖发给量子化学家Walter Kohn教授John Anthony Pople教授,他们的工作使得分子内部的结构更清晰的呈现在我们眼前。2013年诺贝尔化学奖得主Martin Karplus、Michael Levitt和Arieh Warshel为了更加深入研究更为复杂的化学体系,给复杂化学体系设计了多尺度模型,推动了计算化学的发展,这再一次奠定了计算化学举足轻重的江湖地位。
1.4.2量子计算方法
严格意义上来说,我们在对任何一个体系进行分析和计算时都应该秉持科学、严谨、准确的态度,但是求解薛定谔方程的计算量与分子体系的电子数呈指数关系。所以,薛定谔方程的精确求解非常困难,因此需要引入各种近似。对于不同的体系,我们应该采取不同的近似方法,这些近似项有效的简化了薛定谔方程,大大降低了数学求解难度,最终我们可以得到多原子分子体系的数值解。例如,对于单电子类、小分子体系以及大分子体系,计算方法应有不同程度的近似。价键理论是Schrödinger方程近似求解的方法之一,然而它在解释一些问题时存在很多缺陷(如氧分子等顺磁性体系或含大π键体系)。下面列出几种常见的量子化学方法:
- 第一原理方法(也称从头算方法)
从头算方法是指,在做量子计算的过程中近似项不使用实验数据、经验等参数,仅依靠理论推导。第一原理电子结构方法常可分为:
- Hartree-Fock(HF)方法[26]
HF方法是量化中最重要的方法之一,此方法采用了轨道近似,将多电子问题转化为单电子问题从而求解方程。但是这种近似忽略了电子之间的关联项,造成了较大的误差,导致其计算精度不高。HF方程为所有基于分子轨道理论的量化计算方法提供了理论基础,一切的分子轨道计算方法(“Post-Hatree-Fock”、各种半经验计算方法)都是根据HF方法派生得到的。对于稳定的分子和过渡金属处理较好,在处理有孤对电子、共扼、弱键体系时表现较差。
- 后自洽场(SCF)方法
HF方法的误差主要源于它忽略了电子之间的关联项,导致其计算精度不高。为了解决这一问题,后SCF方法开始逐渐发展起来。微扰方法、偶合簇方法、组态相互作用方法等都属于后SCF方法,他们的相同点在于都在HF方法增加电子相关因素,因此脱离了单电子近似,计算结果更加精确。但随着基函数的数量的增加,计算量呈高次幂增长,即使计算机技术已经突飞猛进,这些方法也仅仅适用于一些中小体系的计算。目前常用的后SCF方法主要有:MP2、MP4、 QCISD(T)等。
- 密度泛函理论(Density Functional Therory, DFT)[27-29]
密度泛函理论是以Thomas-Fermi模型为基础,HF方程为依据所提出的。他与HF方法不同的是,密度泛函方法使用电子密度而不是波函数来描述体系的能量,使计算更加简便。相较于后SCF方法,密度泛函方法能在保证计算的精度的基础上更合理使用计算资源,便宜且速度更快。目前常用的密度泛函主要有:B3LYP、bp86、M06-2X、M06-L、ωB97X-D、M05-2X等。
- 半经验计算方法
半经验方法是在求解方程时做了高度近似,其各方面精度和可靠性逊于Hartree-Fock等方法。半经验方法计算速度快,对计算机要求低,经常用于大体系的计算或处理大体系的第一步。例如:AM1、PM3、MNDO。
1.4.3基组
在理论计算中,基组是分子轨道的数学描述,在计算化学当中基组有非常重要的地位。在实际计算中,如何选择合适的基组则尤为重要,因为基组的选用在很大程度上影响着计算结果的精度。下面列出一些常见的基组类型。
- STO-NG基组
STO-NG基组也称为极小机组,或者单zata基组。其中应用组广泛的是STO-3G基组,他是极小机组的首选机组。STO-3G基组小,计算结果的精确程度相对较差,但因其计算量小,常用于试探性寻找反应的过渡态。
- 价层分裂基组
因为化学反应大部分在价轨道发生,为了提高计算的精确程度,对内层轨道用一个STO拟合原子轨道,价轨道用两个STO拟合(内轨与外轨),称为分裂价基。常用的价层分裂基组有:3-21G、4-31G、6-31G、6-311G。
- 极化基组
这种基组针对价层分裂基组无法对电子云性质进行精确描述的问题而引入的,大多数应用于强共轭体系的计算。首先,他允许改变轨道大小,其次他可以改变轨道形状并且增加了角动量。常用的极化基组有:6-31G(d)=6-31G*、6-31G(d,p)=6-31G**。
- 弥散基组
此基组是s和p轨道函数的大号的版本,允许轨道占据更大的空间。适用于计算含有阴离子、电负性高的原子(例如F、Cl)、纯酸的体系等。这种基组是我们计算中最常用到的基组类型。常用的极化基组有:6-31 G(d)、6-31 G(d)。
- 高角动量基组
此基组是在分裂价层基组基础上增加多个极化函数,进一步提高
了计算的精确程度,增强了计算的准确性。常用的高角动量基组有6-311 G(3df,3dp)等。
- 赝势基组
赝势基组用于处理含有第三周期以上的重元素的体系,其中LanL2DZ是最常用的赝势基组。
1.4.4过渡态理论[30-32]
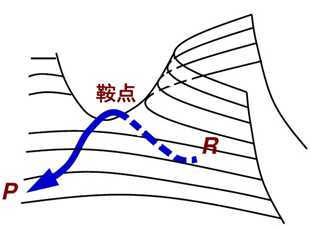
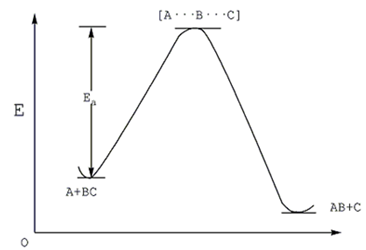
1935年亨利·艾林和迈克尔·波拉尼提出了过渡态理论,该理论认为在有机化学反应中,反应物分子并不仅仅是通过简单碰撞后直接形成产物,而是从反应物到生成物的历程之间有势能较高的活化络合物形成,该状态称为过渡态。过渡态理论又常被称为“绝对反应速率理论”。
如下图所示,反应物A和BC经过活化后,产生了局域势能最高点[A…B…C],该点的能量高于A+BC和AB C,该点结构就是此时反应中过渡态的结构。虽然过渡态确实存在于反应进程中,但它的存在是暂时性的。因为它既不能稳定存在于反应体系中,也不能从整个体系中分离出来。最终,我们可以利用过渡态与反应物和产物之间的活化自由能差、活化焓等等数据来分析计算反应历程。
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: