基于单齿配体策略的邻氯苯甲醛的合成研究毕业论文
2020-02-19 13:39:41
摘 要
邻氯苯甲醛及其衍生物常作为药物中间体,是一种在医药和农药等领域用途非常广泛的化合物。因其应用性很好,邻氯苯甲醛及其衍生物的合成方法被广泛探索。目前,过渡金属催化的碳氢键活化发展成为有机化学中选择性构筑 C-C 键和 C-X 键的最有效工具之一。但C-H键的稳定性以及反应的位点选择性等问题成为相关研究领域关注的焦点,导向基的引入降低了金属参与碳氢键活化的活化能,也在一定程度上提高了反应位点的选择性。由于苯甲醛的丰富性和合成多功能性,以苯甲醛为原料合成邻氯苯甲醛,其合成策略可以适用于多种苯甲醛衍生物的转化,具有重要的应用价值。例如,塞来昔布为原料,将其苯环上的甲基氧化为醛基,即为苯甲醛的衍生物,再进行邻位氯化,实现药物类似物的多样化,使其药理作用更为优良。本实验旨在于应用单齿配体策略合成邻氯苯甲醛及其衍生物。
本实验以苯胺和醛缩合形成的亚胺作为瞬时导向基团,过渡金属催化的醛C(sp2)–H键官能团化实现苯甲醛邻位C–H键的直接官能团化,这种单齿配体策略可以广泛用于苯甲醛的C(sp2)-H官能化的合成方法。首先,本实验采用间溴苯甲醛为底物,N-氯代丁二酰亚胺(NCS)为氯源探究该反应的最优条件。醛的配位能力弱,需加入配体苯胺以形成亚胺,实验中先尝试使用各种苯胺,试出最佳配体,再筛选出配体的用量。随后进行N-氯代丁二酰亚胺用量的筛选、温度的筛选、添加剂的优化、添加剂用量的筛选、溶剂的筛选、各种钯催化剂的筛选。因N-氯代丁二酰亚胺本身就是一种氧化剂,无需另加氧化剂。最后,在最佳反应条件的基础上,我们进行了底物拓展,合成了一系列邻氯苯甲醛衍生物并表征其结构。实验结果表明,大多数底物产率良好,带有给电子或吸电子取代基的苯甲醛均具有良好的耐受性。该反应避免了使用化学计量的试剂和用于安装、移除引导基团的附加步骤,且显著提高位点选择性并扩大底物范围,具有值得更进一步研究的应用潜力。
关键词:苯甲醛;邻氯苯甲醛衍生物;N-氯代丁二酰亚胺;瞬时导向基团
Abstract
O-chlorobenzaldehyde and its derivatives are often used as pharmaceutical intermediates and are a widely used compound in the fields of medicine and pesticides. Because of its good applicability, the synthesis of o-chlorobenzaldehyde and its derivatives has been widely explored.At present, the transition metal-catalyzed activation of carbon-hydrogen bonds has become one of the most effective tools for the selective construction of C-C bonds and C-X bonds in organic chemistry. However, the stability of C-H bonds and the selectivity of the reaction site have become the focus of research in related fields. The introduction of the directing group reduces the activation energy of the metal involved in the activation of carbon-hydrogen bonds, and also improves the selectivity of the reaction site to some extent.Due to the richness and synthetic versatility of benzaldehyde, benzaldehyde is used as raw material to synthesize o-chlorobenzaldehyde. The synthesis strategy can be applied to the conversion of various benzaldehyde derivatives, and has important application value.For example, celecoxib is used as a raw material, and the methyl group on the benzene ring is oxidized to a carbonyl group, which is a derivative of benzaldehyde, and then ortho-chlorination is carried out to realize diversification of the drug analog, thereby making the pharmacological action more excellent.This experiment aimed to synthesize o-chlorobenzaldehyde and its derivatives using the monodentate ligand strategy.
In this experiment, the imine formed by the condensation of aniline and aldehyde is used as thetransient guiding group. The transition metal-catalyzed aldehyde C(sp2)-H bond functionalization realizes the direct functionalization of the ortho-C-H bond of benzaldehyde. This monodentate ligand strategy can be widely used in the synthesis of C(sp2)-H functionalization of benzaldehyde.First, in this experiment, m-bromobenzaldehyde was used as a substrate, and N-chlorosuccinimide (NCS) was used as a chlorine source to explore the optimal conditions for the reaction.The coordination ability of the aldehyde is weak, and the ligand aniline needs to be added to form the imine. In the experiment, various anilines are tried first, and the optimal ligand is tried, and the amount of the ligand is selected.Subsequent screening of N-chlorosuccinimide dosage, temperature screening, additive optimization, additive dosage screening, solvent screening, and screening of various palladium-containing catalysts. Since N-chlorosuccinimide itself is an oxidizing agent, no additional oxidizing agent is required.Finally, based on the optimal reaction conditions, we carried out substrate expansion, synthesized a series of o-chlorobenzaldehyde derivatives and characterized their structure.The experimental results show that most of the substrates have good yields, and the benzaldehydes with electron donating or electron withdrawing substituents are well tolerated. This reaction avoids the use of stoichiometric reagents and additional steps for mounting and removal of the guiding groups, and significantly increases site selectivity and expands the substrate range, with potential applications worthy of further investigation.
Key words: Benzaldehyde; O-chlorobenzaldehyde derivative; N-chlorosuccinimide; Transient guiding group
目录
摘要 I
Abstract II
第1章 绪论 1
1.1 引言 1
1.2 相关药物介绍 1
1.3 瞬时导向基团导向的C-H键活化反应研究现状 4
1.3.1 瞬时导向基团导向的苯甲醛C(sp2)-H键卤化反应 4
1.3.2 瞬时导向基团导向的苯甲醛C(sp2)-H键活化反应 5
1.4 研究目的与意义 8
第2章 邻氯苯甲醛合成的最优条件探索 10
2.1 引言 10
2.2 实验所用试剂、仪器及产物的分析表征 10
2.2.1 试剂 10
2.2.2 仪器 11
2.2.3 分析表征 11
2.3 反应条件的优化 12
2.3.1 配体的筛选 12
2.3.2 胺添加量的优化 13
2.3.3 有机酸的筛选 14
2.3.4 TFA添加量的优化 15
2.3.5 溶剂的筛选 16
2.4 小结 16
第3章 底物适用性探究 18
3.1 引言 18
3.2 实验步骤 18
3.3 底物适用性探究 18
3.3.1 苯甲醛底物范围拓展 18
3.4 小结 20
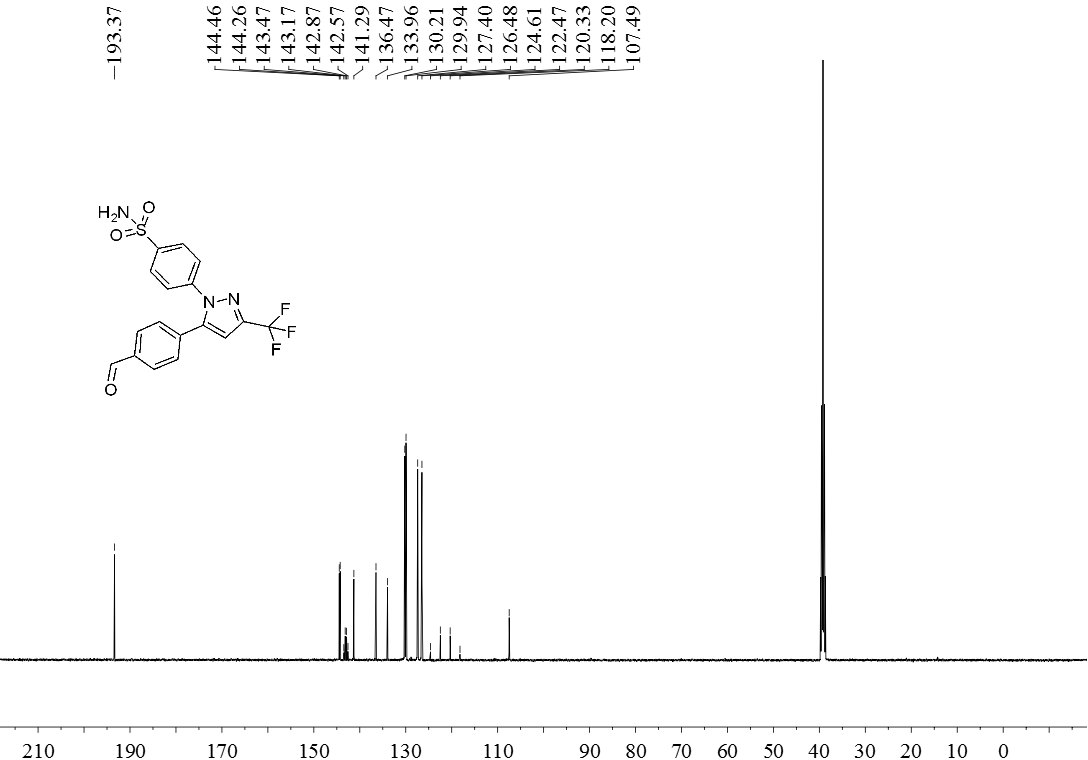
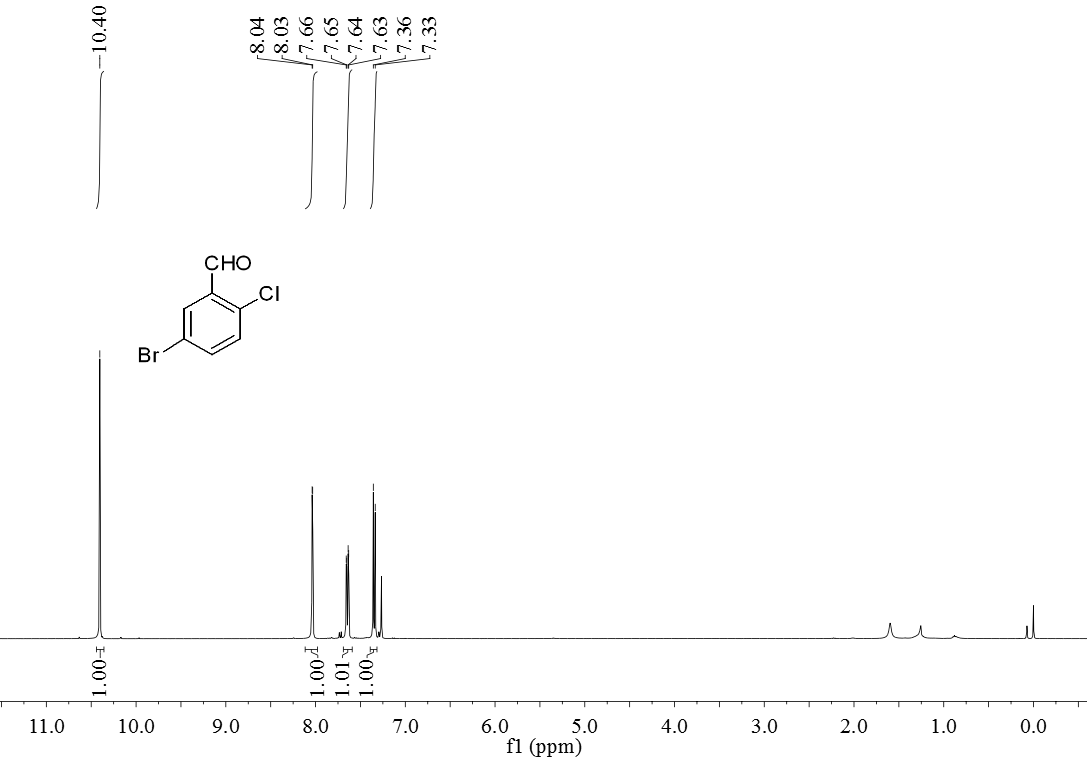
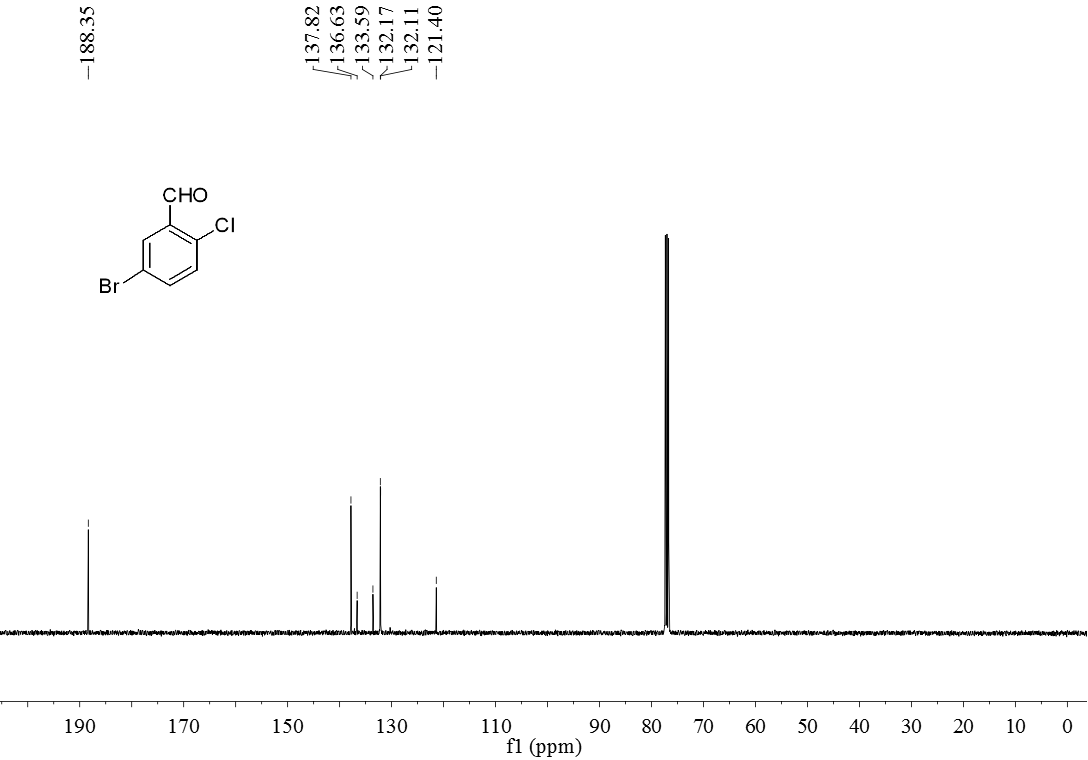
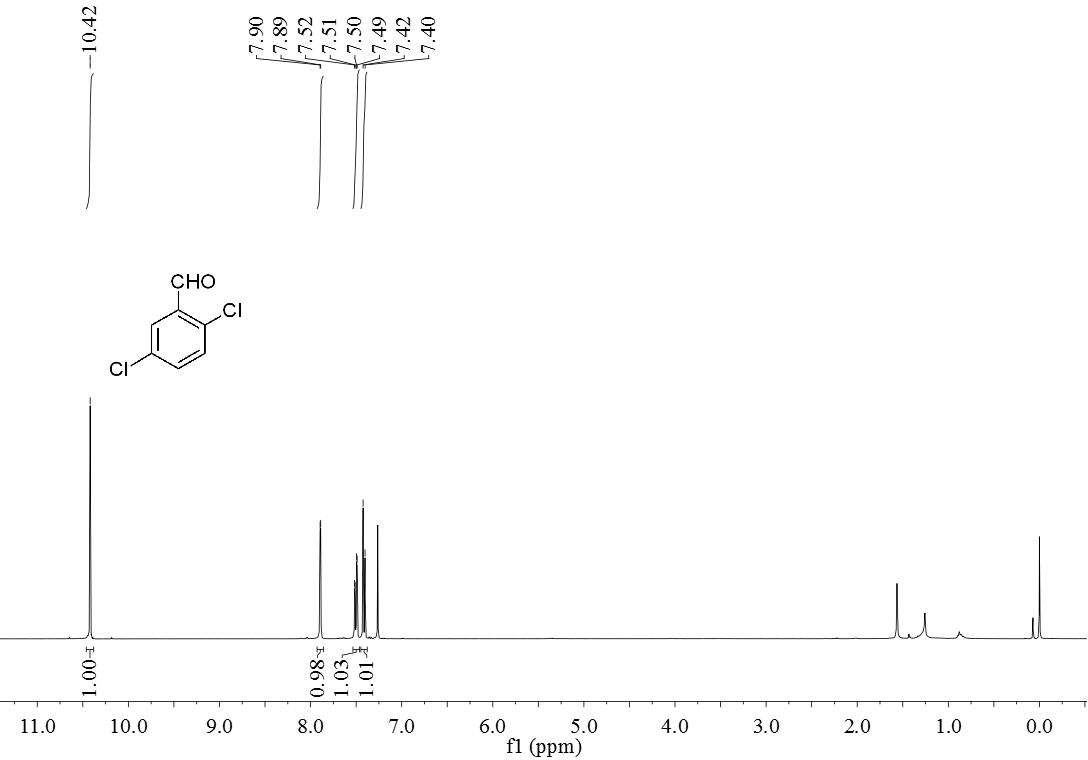
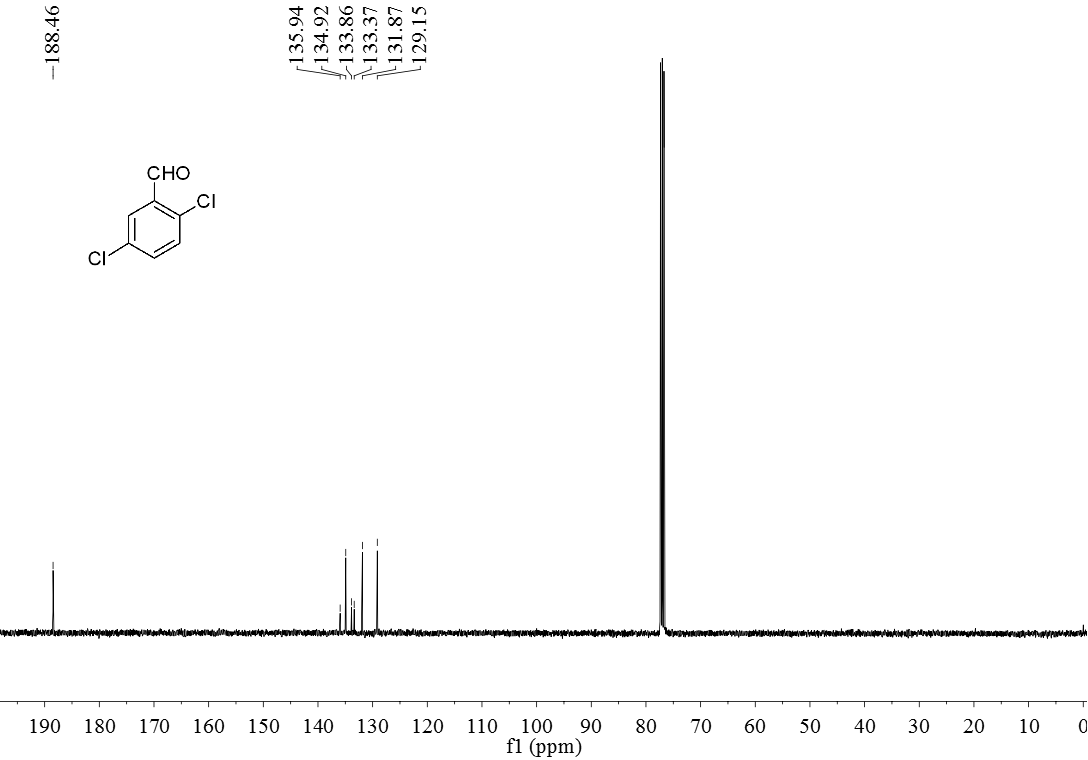
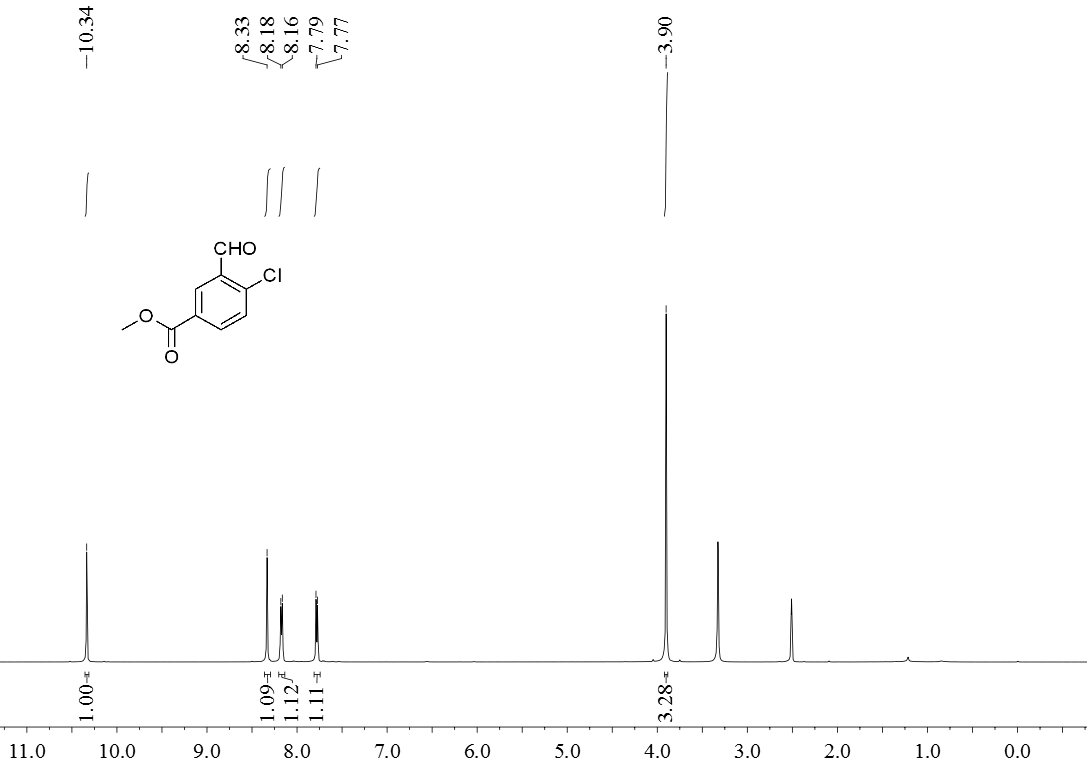
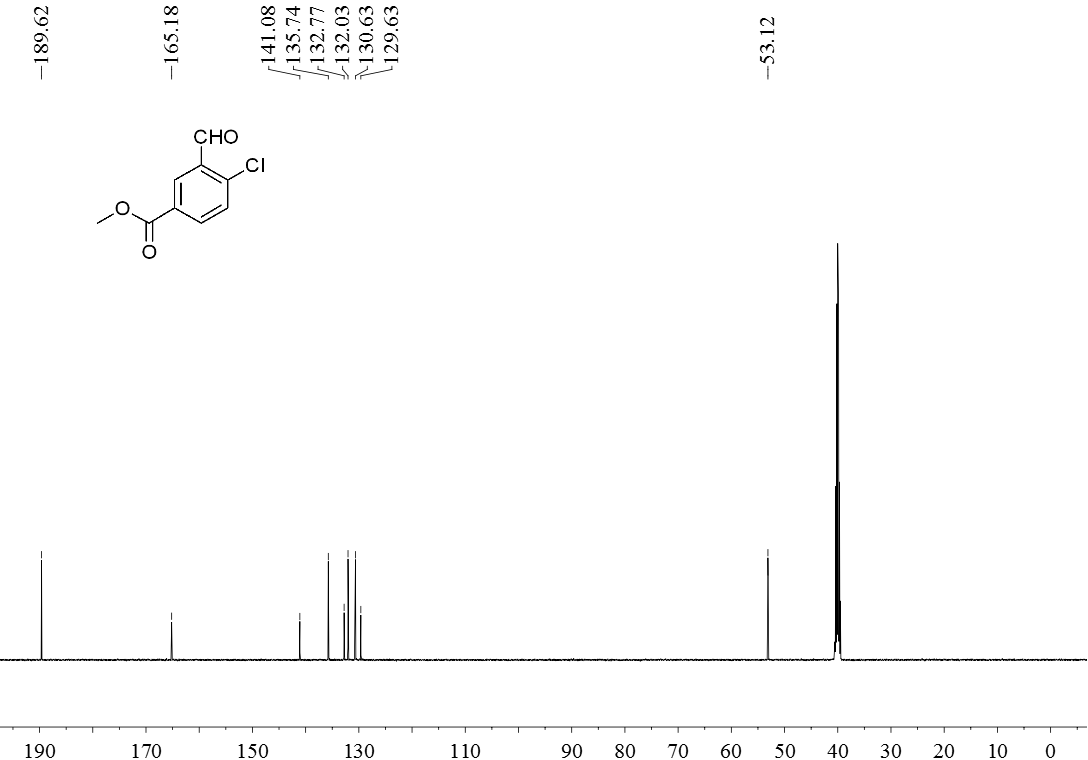
3.5 产物表征数据 21
结论 24
致谢 25
参考文献 26
附录核磁谱图 27
第1章 绪论
1.1 引言
邻氯苯甲醛衍生物是一种在医药和农药等领域被广泛应用的化合物,具有不同的药用性能,可被用于合成氯唑青霉素等医药品,也可用于制造高效杀螨剂。因此,其合成方法受到相关研究领域的极大关注。过渡金属催化C-H键活化合成C-X键、C-C键等以实现化合物功能化的方法是一种新兴的合成策略,将其应用于邻氯苯甲醛类药物的合成,可以简化其合成路线。
醛基的导向作用很弱,这是由于醛基的弱协调性,对氧化剂的敏感性,以及不允许金属插入到C-H键中的属性,阻碍了醛基作为导向基团在C-H键活化领域的发展。同时,转化的范围受到限制,因为优先激活在醛基C-H键上的惰性芳基C-H键,其可以进行无金属氧化或金属插入。为了解决这个问题,双齿配体策略利用氨基酸可逆地与醛或酮在原位缩合形成亚胺,以用于活化芳基化合物邻位的惰性C–H键。Zhang课题组[1]在实现钯催化苯甲醛邻位 C(sp2)-H 芳基化反应后,随之尝试了苯甲醛邻位 C(sp2)-H 键的氯化及溴化反应。该实验中,以钯为催化剂,以邻氨基苯甲酸类化合物与醛缩合形成的亚胺作为瞬时导向基团,以TFA为添加剂,以DCE为溶剂,以NCS(NBS)为氧化剂和氯(溴)源,含有各种取代基(如砜基,酯基,氨基甲酸酯,氰基,酰胺等)的苯甲醛为底物,都可以得到良好的产率。醛基可逆地与胺在原位缩合形成亚胺作为瞬时导向基团优于能够导向C-H键活化的其他配位官能团,从而显著扩大过渡金属催化C-H键官能化的底物范围并提高了反应位点的选择性。本实验以苯胺替换氨基酸,将单齿配体策略应用于邻氯苯甲醛及其衍生物的合成,简化了配体胺的结构,使反应更易于进行。这种方法不仅可以用于简化邻氯苯甲醛类药物的合成路线,也可以用于药物类似物的多样化研究。
1.2 相关药物介绍
氯唑青霉素[2]是抑制细菌细胞壁合成的β-内酰胺类药物,是临床上被广泛使用的一种抗生素。此药物具有噻唑环和β-内酰胺环两个环,6-氨基青霉烷酸为其母核结构。氯唑青霉素是一种半合成的异恶唑类耐酶青霉素,对青霉素酶稳定,抗菌谱与青霉素类似,对耐青霉素的金黄色葡萄球菌有一定作用且作用效果比苯唑西林要强,对青霉素敏感的阳性球菌的抗菌作用不如青霉素有效。可以口服,也可注射给药,用于表皮葡萄球菌和金黄色葡萄球菌的周围感染,一般不适用于中枢感染,主要用于耐青霉素葡萄球菌所致的各种感染,如心内膜炎、骨髓炎、呼吸道感染、软组织感染、脓毒症、烧伤、脑膜炎等,也可用于耐青霉素金黄色葡萄球菌与化脓性链球菌或肺炎链球菌所致的混合感染。其不良反应与青霉素相似,较为轻和,毒性很低,一般情况能充分耐受,最常见的为肠胃反应,如呕吐、恶心等,常见的有过敏性反应,如药疹、过敏性休克、溶血性贫血、粒细胞减少、血清病型反应等。为防止过敏反应的发生,用药时应询问病史,包括药物过敏史、家属过敏史、用药史,并要进行皮肤过敏试验。不可空腹用药或皮试,为发生过敏性休克时能及时治疗,应做好急救准备,如氢化可的松、肾上腺素、等药物及注射器材。大剂量注射时要监测血清离子浓度,以防高钠血症的发生。其结构式如下:
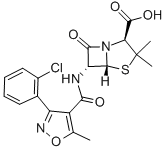
图1.1氯唑青霉素结构式
塞来昔布[2]是一种解热镇痛抗炎药,也称作非甾体类抗炎药(NSAIDs),具有解热、镇痛、抗风湿、抗炎作用。环氧酶(COX)至少有固有型环氧酶(COX-1)和诱生型环氧酶(COX-2)两种同工酶,塞来昔布属高选择性COX-2抑制剂。炎症损伤主要刺激内皮细胞、血管平滑肌、巨噬细胞、单核细胞、成纤维细胞等,诱导诱生型环加氧酶的生成,诱生型环加氧酶是触发后续炎症反应的关键环节。炎症反应的中心环节为血管反应,局部反应为功能障碍及红、肿、热、痛等,全身反应为末梢血中性粒细胞升高及发热。研究发现,COX-2对于保护肾脏及血管内皮细胞合成PGI2(前列环素)具有重要意义,高选择性COX-2抑制剂可能增加高血压和水肿的发病率。塞来昔布与传统的非甾体类抗炎药相比,具有相同的疗效,即解热、镇痛、抗炎等作用,同时显著降低了药物的胃肠道不良反应,如溃疡、出血等,但对患者的肾毒作用依然存在,且增大了心血管不良反应的风险。在常用剂量下,对血小板COX没有抑制作用。口服吸收良好,经肝脏CYP2C9代谢,可抑制CYP2D6的活性,因此,与影响CYP2D6和CYP2C9活性或经这两种酶代谢的药物合用时,要注意它们在代谢方面的相互作用。在临床上,塞来昔布主要用于原发性痛经、急性疼痛、强直性脊椎炎、风湿性关节炎、骨关节炎、类风湿性关节炎等。临床使用此药物时,应遵循最短疗程和最小有效量的原则,不推荐作为非甾体类抗炎药的首选药。常见的不良反应为消化不良、腹泻及上腹疼痛。阿司匹林等其他非甾体类抗炎药及磺胺类药物过敏者禁用。截至2012年,仅有美洛昔康和塞来昔布这两种高选择性COX-2抑制剂被美国批准上市。因此,在保留甚至增强药效并减少毒副作用的基础上,实现此药物类似物的多样化具有重要现实意义。其结构式及反应式[4]如下:
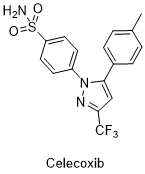
图1.2塞来昔布结构式
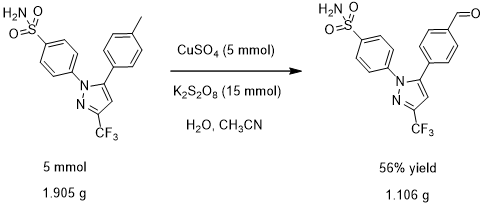
图1.3塞来昔布被氧化为苯甲醛衍生物的反应式
实验室合成塞来昔布衍生物的过程:将1.150 g(5 mmol)硫酸铜五水合物和4.055 g(15 mmol)过硫酸钾溶于40 mL水中,得到蓝色混合溶液。然后,将混合溶液加热至65℃; 再将1.905 g(5 mmol)塞来昔布溶解在20 mL乙腈溶剂中,并加入到混合溶液中,得到浅绿色溶液。将浅绿色溶液加热回流45分钟; 通过TLC跟踪反应。反应完成后,将水和二氯甲烷加入到反应混合物中,并用二氯甲烷萃取3次。将合并的有机层用无水硫酸钠干燥,并真空浓缩。通过硅胶柱色谱法纯化粗产物,得到期望的纯净产物,为白色固体(1.106 g,56%)。
1.3 瞬时导向基团导向的C-H键活化反应研究现状
1.3.1 瞬时导向基团导向的苯甲醛C(sp2)-H键卤化反应
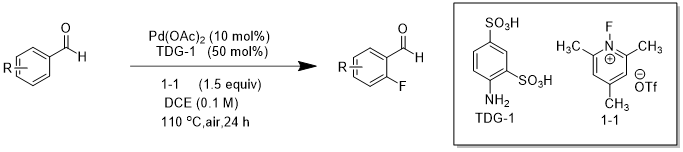
图1.4钯催化的苯甲醛邻位C(sp2)-H键氟化反应
2018 年,Sorensen课题组[3]使用新型的瞬时引导基团邻氨基苯磺酸(TDG-1),在醋酸钯催化下,实现了苯甲醛的邻位 C(sp2)-H 的直接氟化(图 1.4)。在该实验中,Sorensen 课题组以1-氟-2,4,6-三甲基吡啶三氟甲磺酸酯(1-1)为反应中的氟源和亲电子氟化剂。反应最佳温度为110 ℃,最适宜的反应时长为24 h。经过多种溶剂的筛选,结果表明DCE 为最优反应溶剂,然而在此种情况下,会有少量苯甲醛的氯化物产生。Sorensen 课题组在尝试使用传统的瞬时导向基团邻氨基苯甲酸(TDG-2)时几乎没有目标产物生成。苯甲醛钯化合物中间体的X射线晶体结构在稳定配体稳定剂三苯基膦作用下可获得。
以上是毕业论文大纲或资料介绍,该课题完整毕业论文、开题报告、任务书、程序设计、图纸设计等资料请添加微信获取,微信号:bysjorg。
相关图片展示: