用于太阳能电池应用的有机增敏剂的分子工程外文翻译资料
2022-11-19 14:24:41

英语原文共 7 页,剩余内容已隐藏,支付完成后下载完整资料
用于太阳能电池应用的有机增敏剂的分子工程
摘要:新型有机敏化剂包括供体、电子导电,锚固基团被设计在分子水平和合成。官能化的不对称有机敏化剂3 { 5 - [(N,N-二9,9-dimethylfluorene-2-yl)苯基] - thiophene-2-yl } - 2-cyano-acrylic酸(饱和)和3 { 5′- [ N,N-二(9,9-dimethylfluorene-2-yl)苯基] - 2′- bisthiophene-5-yl } - 2-cyano-acrylic酸(JK-2),在锚定到TiO2薄膜,表现出前所未有的入射光子的电流转换效率91%。光伏数据使用一个电解质组成的0.6 M-methyl-N-butyl imidiazolium碘,碘0.04,0.025 LiI,硫氰酸胍盐,0.05米和0.05米tert-butylpyridine 15/85(v / v)戊腈和乙腈的混合物显示短路光电流密度14.0plusmn;0.2 mA / cm2,开路电压753plusmn;10 mV,填充因数为0.76plusmn;0.02,对应于一个整体转换效率8.01%的标准是1.5下的阳光。DFT/TDDFT的计算已经在这两个有机感光器上进行,以了解它们的结构、电子和光学性质。我们的研究结果表明,氰基丙烯酸基团在噻吩单位上基本是共面的,反映了噻吩-氰基丙烯酸基团的强共轭性。分子轨道分析证实了氧化还原电位的实验分配,而TDDFT计算则允许分配可见吸收带。
染料敏化太阳能电池(DSCs)作为传统固态光伏器件的低成本替代品引起了广泛关注。这些电池主要使用钌基聚吡啶配合物作为电荷转移增敏剂,在标准的全球空气质量1.5阳光下,产生超过11%的太阳能-电能转换效率。为了扩大DSC这一关键部分的选择,几个小组已经开发出了不含金属的增敏剂,并在5-8%的范围内获得了效率。有几个基本的要求,指导分子工程的高效敏化剂。激发态氧化还原电势应与氧化物导电带边缘的能量相匹配。光的激发应该与从染料的光收集到表面的矢量电子流动相联系,从而提供从激发染料到TiO2传导带的有效电子传递。最后,在染料和TiO2传导带之间,通过供体和锚定组和良好的电子耦合的强烈共轭,以保证高的电子传递速率。
DSC中许多有机染料的低转换效率的一个主要因素是在半导体表面形成的染料聚集物。这种聚集现象会影响滤光效应的光吸收。因此,为了获得最佳性能,需要通过适当的结构修饰来避免有机染料的聚集。有机染料的另一个重要问题是它们的稳定性,通常比金属复合物的稳定性要低,很可能是由兴奋的三重态和不稳定的激发态形成的出血通道。
图1所示。通过DFT计算,优化了JK-1和JK-2的分子结构(详见文本)。原子标签也被报道。
为了结合这些需要的特性,我们设计并合成了新型的非对称有机增敏剂JK-1和JK-2(见图1),其中包括双二甲基氟烯苯胺作为电子供体和氰基丙烯酸的作用作为受体,这两种功能通过导电噻吩单元连接。在JK-1和JK-2增敏剂中定制的二甲基氟苯胺的细微差别,确保在暴露于光照和高温下,与简单的芳基胺相比,耐降解性更强。另外,二甲基芴的非平面结构抑制了聚集,不利于分子堆积。最后,桥接噻吩单元用于提供供体与锚定组之间的接合,并增加染料的摩尔消光系数。在此,我们报告两种感光剂JK-1和JK-2的合成、表征和光电性能。此外,我们进行DFT/TDDFT计算,提供了两种增敏剂的结构、电子和光学性质的详细描述。
结果与讨论
功能化不对称的增敏剂3 - { 5 -[N,N-bis(9,9-dimethylfluorene-2-yl)苯基]-thiophene-2-yl } 2-cyanoacrylic酸(JK-1)和3 - { 5′-(N,N-bis(9,9-dimethylfluorene-2 - yl)苯基]2,2′-bisthiophene-5-yl } 2-cyano-acrylic酸(JK-2)方便地通过反应合成5 -(N,N-bis(9,9 - dimethylfluorene-2-yl)苯基]-thiophene-2-carbaldehyde和3 - { 5′-(N,N-bis(9,9-dimethylfluorene-2-yl)苯基]2,2′- bisthiophene-5-yl } 5-carbaldehyde与氰乙酸乙腈的哌啶,分别(有关详细信息,请参阅合成部分)。
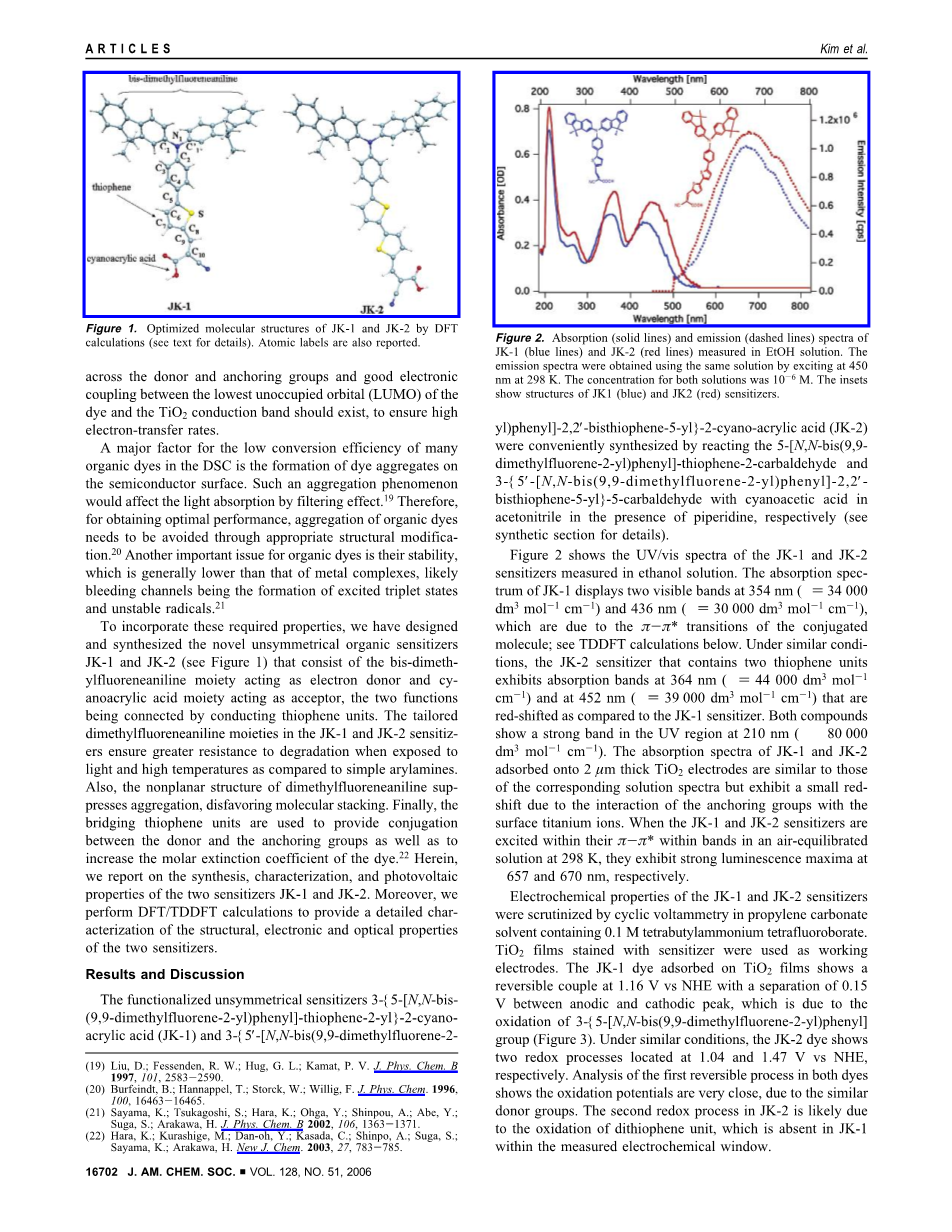
图2。在EtOH溶液中测量JK-1(蓝线)和JK-2(红线)的吸收(实线)和发射(虚线)光谱。利用同样的溶液,在298k的450nm处获得了发射光谱。两种溶液的浓度为10-6 m, insets显示JK1(蓝色)和JK2(红色)增敏剂的结构。
图2显示了在乙醇溶液中测量的JK-1和JK-2增敏剂的UV/vis光谱。JK-1的吸收光谱显示了在354 nm () 354 nm () 340dm3 mol1 - cm-1)和436nm () 30000dm3 mol- l-1 cm-1的两个可见波段,这是由于共轭分子的变化;请参阅下面的TDDFT计算。在类似的条件下,包含两个噻吩单位的JK-2增敏剂在364 nm () 44 000 dm3 mol1 -1 cm-1)和452 nm () 39000 dm3 mol-1 - cm-1的吸收波段,与JK-1敏化剂相比是红移的。两种化合物显示出强大的乐队在210 nm紫外线地区(asymp;80 000 dm3 mol-1 cm - 1)。JK-1和JK-2吸附在2 m厚TiO2电极上的吸收光谱与相应的溶液光谱相似,但由于锚定基团与表面钛离子的相互作用,呈现出小红移。当JK-1 JK-2增敏剂兴奋在pi;-pi;*在乐队在298 K air-equilibrated解决方案,他们表现出很强的发光maximasim;657和670nm,分别。
图3。循环伏安法的JK-1(顶面板)和JK-2增敏剂(底板)附加到纳米晶二氧化钛薄膜沉积在导电玻璃FTO(电极尺寸0.28平方厘米,膜厚度2.5micro;m颗粒大小20nm)。这个薄膜是在450°C下烧制 20分钟。10分钟冷却后,在染料溶液中沉浸一夜。用纯乙腈清洗敏感电极,并在无惰性气氛下以100 mV s-1的扫描速度在手套箱内测量。
摘要采用循环伏安法研究了含0.1 M四氟铵四氟硼酸丙烯酯溶剂的JK-1和JK-2增敏剂的电化学性能。用增感剂染色的TiO2膜作为工作电极。JK-1染料吸附在二氧化钛薄膜展示了一个可逆的结合在1.16 V和NHE阳极和阴极峰之间的间隔0.15 V,这是由于氧化3 - { 5 -[N,N-bis(9,9-dimethylfluorene-2-yl)苯基]组(图3)。在相似的条件下,JK-2染料显示两个氧化还原过程位于1.04和1.47 V vsNHE,分别。两种染料的第一个可逆过程的分析表明,由于类似的供体群,氧化电位非常接近。在JK-2中第二个氧化还原过程可能是由于dithiophene单元的氧化,该装置在测量的电化学窗内没有JK-1。
图4。JK-1(蓝线)和JK-2(红线)的循环伏安图,在含有0.1 M TBA(PF6)的DMF溶液中,使用玻璃碳作为一个工作和一个Pt计数器电极,扫描速率为100 mV s-1。
图4为DMF溶液中JK-1和JK-2的循环伏安图,使用0.1 M的四丁基四溴铵作为支撑电解质和玻璃碳工作电极。这两种染料在1.06和1.01 V对NHE中都有一个准可逆波,它们被重新分配到3-{5-[N,N-bis(9,9-二甲基芴- 2-yl)的氧化。对负电位扫描时,两个增敏剂显示quasi-reversible波E1/2)-1.34和-1.30 V,分别(见图4)。第一个还原电位的积极转变JK-2 JK-1相比,是由于pi;-conjugation的延伸,符合下面给出的理论分析。
为了深入了解JK-1和JK-2染料的几何、电子和光学特性,我们对JK-1的两种有机增敏剂和时间依赖性DFT (TDDFT)计算进行了DFT计算,使用了Gaussian 03程序包。特别是,我们使用了B3LYP交换相关的functional26和一个6-31g*基集;27个溶剂效应被包含在可极化连续模型中。28我们优化了气相JK-1和JK-2的分子结构,没有任何对称约束,得到图1所示的几何图形。值得注意的是,JK-1和JK-2优化结构计算两个dimethyl-fluorene配体绑定到苯胺N1,安排出平面的方式(见图1)最小化位阻,所反映的往上平移的计算值(C′1)-N1-C2-C3二面角角度31.6 - -31.4°和34.1 - -33.7°,分别为JK-1和JK-2。苯胺和噻吩的飞机之间形成的角度计算是20.5°和22.7°JK-1 JK-2复合物,分别虽然cyanoacrylic酸组被发现本质上是对噻吩单元共面,反映出强烈的接合在thiophene-cyanoacrylate组。我们还注意到,在JK-2的情况下,两个噻吩单位本质上是共面的,它们都考虑到两个配体的cisoid和transoid的排列,后者只受到0.7 kcal/mol的支持。
图5。在真空和乙醇中,JK-1和JK-2的前沿分子轨道的示意图。
然后我们分析了在真空和乙醇溶液中有机感光剂的电子结构,这是用来记录实验光谱的溶剂。分子轨道能量的示意图如图5所示,图6中我们报道了JK-1前沿分子轨道的等密度图。最高占据分子轨道(HOMO)的两个增敏剂,在真空和解决方案,是离域bis-dimethyl-fluoreneaniline配体,与最大组件产生的氮孤对周围的配体和pi;框架;HOMO-1是,另一方面,pi;-bonding轨道离域在整个分子,与最大组件噻吩和cyanoacrylic半个;参见图6。最低未占据分子轨道(LUMO)的增敏剂pi;*轨道离域的噻吩和cyanoacrylic组,与巨大的组件从青色和羧基的根;LUMO 1 =pi;*轨道的两个dimethylfluorene配体;参见图6。
图6。JK-1的HOMO-1、HOMO、LUMO和LUMO 1的等密度表面积。类似的轨道是用JK-2计算的。
对JK-1和JK-2的边界轨道的检验表明,这两个物种的HOMO和LUMO本质上是能量上的孤立轨道,它们分别位于HOMO和LUMO之间,分别位于HOMO和LUMO之间。值得注意的是,JK-2的HOMO和LUMO与对应的JK-1轨道相比,分别被稳定和稳定了0.10和0.16 eV。因此,JK-2 HOMO-LUMO的间隙减小到2.21 eV,而JK-1则为2.47 eV,由于在配体上的共轭量增加,因此具有相当大的LUMO稳定性,这是由于第二次的thiophene单元。溶化效应的加入并不会导致上述电子结构图发生质的变化,即使计算相对于气相比较小的同lumo裂隙,主要是因为在溶液中发生的HOMO不稳定;参见图5。
将我们计算的电子结构与现有研究中获得的电化学和光谱数据进行比较是值得的。我们注意到计算的同lumo间隙的趋势与光谱数据很好地比较,显示从JK-1到JK-2的吸收最大值的红移。同时,电化学氧化和还原电位数据符合趋势的HOMO和LUMO能量计算两个染料,显示增加(减少)的人类(LUMO)能量从JK-1 JK-2,很好地与小负(正)的变化测量氧化(还原)的过程。
为了深入了解激发态,产生了两种增敏剂的强吸收光谱,我们在B3LYP/6-31G*水平上进行了TDDFT激发态的计算。考虑到溶剂对电子结构的影响可以忽略不计,TDDFT的计算是在真空中进行的,并且仅限于JK-1。在TDDFT计算中,计算了最低的10个单线态激发,达到了约4 eV (310 nm)的能量。在考虑的能量范围内,我们计算了3个大强度的跃迁(f gt; 0.1),其能量、振子强度和分子轨道贡献的组成均在表1中。
最低的跃迁是在2.23 eV,对应于从双-二甲基芴基的HOMO到LUMO的电荷转移(CT)激发,定位于噻吩-氰基丙烯酸盐的部分。相对于实验吸收最大值,在2.84 eV,计算的跃迁是显著的红移。这与此转换的扩展的电荷转移特性有关,而TDDFT计算并没有使用当前的交换相关函数来正确地捕获它。实验发现在3.50 eV(354nm)似乎是由两个几乎重叠pi;-pi;*转换不同的角色,在3.20和3.37 eV计算,(1)和(2)在表2中,分别。在3.20 eV中计算的跃迁是一个从HOMO-1到LUMO的激发态,因此在噻吩-氰基丙烯酸盐中发生。稍微不那么激烈过渡计算3.37 eV,另一方面,pi;-pi;*激发人类的LUMO 1,因此,相应的励磁bis-dimethylfluoreneaniline中的一部分。更好的协议的计算和实验吸收能量pi;-pi;*特性比CT激发有关pi;-pi;*的本地化特征作用,涉及大幅重叠的轨道。
增敏剂的激发态氧化电位在电子注入过程中起着重要的作用。忽略了光吸收过程中的熵变,根据eq 1,可以从基态氧化偶和零激发能量E(0-0)中得到。
E(S /S*) ) E(S /S) - E(0-0) (1)
从发射光谱中,分别提取了2.43和2.37 eV的E(0-0)能量,分别为JK-2提取。两种增敏剂JK-1和JK-2的激发态氧化电位分别为-1.27和-1.36 V对NHE,明显比N719染料(-0.98 V对NHE)和TiO2传导带的等效电位更低。因此,这种类型的增感剂可能变得非常有吸引力,特别是对具有费米能级的半导体材料来说,其比TiO2的负性更大,因为在导电带和氧化还原电偶之间的间隙增大,会导致更高的开路电位,大大提高了电池的效率。另一方面,两个增敏剂的氧化电位比我更加积极——/ I3 -氧化还原电对(0.4sim;V vsNHE),确保有足够的驱动力的染料再生反应有效地竞争与夺回注入电子的染料阳离子自由基。
增敏剂被用来制造太阳能电池设备探讨电流电压特性使用10 4micro;m二氧化钛透明层。丝网印刷双层膜,由10micro;m透明层和4micro;m散射层,是准备和处理40毫米四氯化钛溶液使用之前报告的过程。13日,23日的二
剩余内容已隐藏,支付完成后下载完整资料
资料编号:[23720],资料为PDF文档或Word文档,PDF文档可免费转换为Word



